
1,3-dipolární cykloadice
1,3-dipolární cykloadice je chemická reakce mezi 1,3-dipólem a dipolarofilní sloučeninou, při níž vzniká pětičlenný cyklus. První takové reakce byly provedeny na přelomu 19. a 20. století, krátce po objevu 1,3-dipólů. Mechanismus a využití těchto reakcí v organické syntéze byly zjištěny v 60. letech 20. století, a to hlavně díky pracím Rolfa Huisgena.[1] Reakce tak někdy bývá označována jako Huisgenova cykloadice (tento název se používá zejména pro 1,3-dipolární cykloadiční reakce organického azidu s alkynem za vzniku 1,2,3-triazolu). 1,3-dipolární cykloadice je významnou metodou regio- a stereoselektivní přípravy pětičlenných heterocyklů a jejich acylových derivátů s otevřenými řetězci.

Mechanismus
Původně byly navrženy dva mechanismy 1,3-dipolární cykloadice: prvním je pericyklický cykloadiční mechanismus, navržený Rolfem Huisgenem;[2] druhým pak postupný mechanismus zahrnující biradikálový meziprodukt, jejž navrhl R. Firestone.[3] Po mnoha debatách je nyní víceméně přijímán první mechanismus,[4] kde 1,3-dipól reaguje s dipolarofilem ve spojené, často asynchronní, reakci a symetrií umožněné π4s + π2s produkty přes šestielektronový aromatický meziprodukt. Existuje ovšem několik příkladů postupného mechanismu nekatalytických 1,3-dipolárních cykloadičných reakcí u thiokarbonylylidů[5] a nitriloxidů[6]

Pericyklický mechanismus
Huisgen zkoumal řadu cykloadičních reakcí 1,3-dipolárních diazosloučenin s nejrůznějšími dipolarofilními alkeny.[2] Následující zjištění podporují spojený pericyklický mechanismus a vyvracejí postupný biradikálový nebo polární mechanismus:
- Vliv substituentů: Různé substituenty na dipólu nevykazují výrazný vliv na rychlost cykloadice, takže při reakci nevzniká nábojem izolovaný meziprodukt.
- Vliv rozpouštědel: Polarita použitého rozpouštědla jen málo ovlivňuje rychlost cykloadiční reakce, což je v souladu s pericyklickým mechanismem, kde polarita nezpůsobuje výrazné změny při přeměně reaktantů na přechodný stav.
- Stereochemie: 1,3-dipolární cykloadice jsou vždy stereospecifické s ohledem na dipolarofil (například z cis-alkenů vždy vznikají syn-produkty), což opět podporuje pericyklický mechanismus, kde se dvě vazby sigma tvoří současně.
- Termodynamické parametry: 1,3-dipolární cykloadice mají neobvykle vysoké záporné hodnoty entropie aktivace, podobně jako Dielsova–Alderova reakce, což naznačuje, že je přechodný stav velmi uspořádaný, a rovněž souhlasí s pericyklickým mechanismem.
1,3-dipól

1,3-dipól je organická molekula, která se může vyskytovat ve zwitteriontové oktetové/sextetové struktuře allylového i propargylového/allenylového typu. Oba tyto druhy 1,3-dipólů sdílejí čtyři elektrony v π-systému nad třemi atomy. Allylový typ je zakřivený zatímco propargylový/allenylový typ má lineární molekulární geometrii.[7] Jsou známy i 1,3-dipóly obsahující prvky vyšších řad periodické tabulky, jako jsou síra a fosfor, ovšem ty nejsou tak často využívány.
Rezonanční struktury lze zobrazit jako delokalizované kladné i záporné náboje na obou koncích dipólu (viz následující schéma). Přesnější metody pro popis rozdělení elektronů u 1,3-dipólů, jako například měření dipólového momentu, jsou založeny na experimentálních či teoretických datech[8] či výpočtech.[9] Například diazomethan má největší záporný náboj na koncovém atomu dusíku, zatímco azoimid má největší záporný náboj na prostředním atomu dusíku.

V souvislosti s tím mohou konce molekul 1,3-dipólů reagovat s nukleofilními i elektrofilními činidly najednou. Nukleofilnost a elektrofilnost každého konce lze zjistit využitím hraničních molekulových orbitalů, které lze získat s použitím výpočetní techniky. Obecně lze říci, že stom s největším orbitalovým koeficientem v rámci HOMO funguje jako nukleofil, zatímco atom v LUMO funguje jako elektrofil. Nejvíce nukleofilním atomem je obvykle, ne však pokaždé, ten, na němž je největší elektronová hustota.[10][11][12] U 1,3-dipolárních cykloadicí je identita páru dipól-dipolarofil určena tím, zda převažuje HOMO nebo LUMO charakter dipólu.
Dipolarofil
Nejčastěji používanými dipolarofily jsou alkeny a alkyny. Dipolarofily obsahující heteroatomy jako jsou karbonylové sloučeniny a iminy mohou také podstoupit 1,3-dipolární cykloadici. Dalšími příklady dipolarofilů jsou fullereny a nanotrubice, u nichž může proběhnout 1,3-dipolární cykloadice s azomethinylidy v Pratově reakci.
Stereospecifita
1,3-dipolární cykloadice často vyústí v „zadržení konfigurace“ s ohledem na 1,3-dipól a dipolarofil. Vysoká stereospecifita silně podporuje spojený reakční mechanismus oproti postupnému, při němž by byla nízká až nulová.
Vliv dipolarofilu
cis-substituenty na dipolarofilním alkenu vytváří cis- a trans-substituenty vytváří trans strukturu výsledné sloučeniny s pětičlenným cyklem.[13]

Vliv dipólu
Stereochemie dipólu obecně není objektem velkého zájmu, jelikož pouze několik dipólů může vytvořit stereogenní centra a rezonanční struktury umožňují rotaci vazby, čímž dochází k narušení stereochemie. Při studii azomethinylidů bylo však ověřeno, že stereospecifitu 1,3-dipolární cykloadice ovlivňují i dipóly. Diastereomerně čisté azomethinylidy se připraví elektrocyklickým otevřením cyklu aziridinů a následně se okamžitě zachytí silnými dipolarofily dříve, než může dojít k rotacím vazeb (viz níže uvedený obrázek).[14][15] Při použití slabších dipolarofilů mají vazby na dipólu dostatek času k rotaci a dojde tak k nespárované stereospecifitě cykloadice.
Tyto výsledky potvrzují, že 1,3-dipolární cykloadice je stereospecifická a že stereospecifitu ovlivňují 1,3-dipól i dipolarofil.
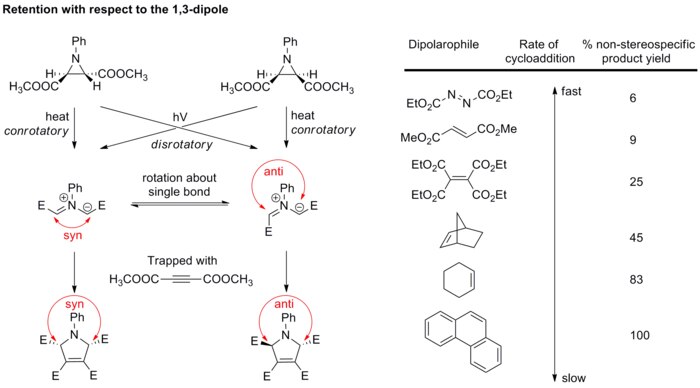
Diastereospecifita
Když se během reakce vytvoří dvě nebo více chirálních center, lze zíksat diastereomerní přechodné stavy a produkty. V Dielsově–Alderově cykloadici je obvykle pozorována endo diastereoselektivita, což je způsobeno sekundárními orbitalovými interakcemi. U 1,3-dipolárních cykloadicí ovšem diastereoselektivitu ovlivňují dve síly: přitažlivé π interakce (podobné sekundárním orbitalovým interakcím u Dielsovy–Alderovy reakce) a odpudivé sterické interakce. Tato působení se často navzájem vyruší, což vede k nízké diastereoselektivitě 1,3-dipolárních cykloadicí.
Příklady substrátem řízené diastereoselektivních 1,3-dipolárních cykloadicí jsou zobrazeny níže. Prvním je reakce benzonitrilu N-benzylidu a methylakrylátu. V přechodných stavech se fenylová a methylesterová skupina spojí tak, že dojde k cis substituci za vzniku výsledného pyrrolinového produktu. Upřednostňovaná π interakce vyváží vzájemné sterické odpuzování fenylové a methylesterové skupiny.[16] Druhým je reakce nitronu a dihydrofuranu. Dosažením exo selektivity se omezí sterické odpuzování.[17] Poslední případ představuje vnitromolekulární reakce azomethinového ylidu s alkenem. Diastereoselektivita je ovládána tvorbou méně narušovaného cis systému spojených cyklů.[18]

Řízená 1,3-dipolární cykloadice
Cykloadici lze ovládat a dosáhnout tak diastereoselektivní reakce. Například kovové ionty mohou být chelatovány dipolarofilem a dipólem a řídit tak reakci selektivně. Níže uvedený obrázek zobrazuje adici nitriloxidu na enantiomerně čistý allylalkohol za přítomnosti hořečnatých kationtů. Nejstabilnější konformace alkenu je ta, u níž se hydtroxylová skupina nachází nad rovinou alkenu. Hořčík je následně chelatován hydroxylovou skupinou a kyslíkovým atomem nitriloxidu. Cykloadice tedy probíhá selektivně.[19]
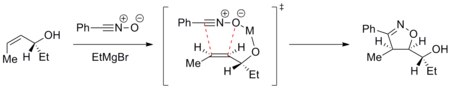
Metody diastereoselekce se využívají například při syntéze epothilonů.[20]

Regioselektivita
U asymetrických párů dipól-dipolarofil mohou vzniknout dva regioizomerní produkty. Regioselektivitu 1,3-dipolárních cykloadicí ovlivňují jak elektronové/stereoelektronové jevy, tak i sterické faktory.[21]
Elektronové/stereoelektronové jevy
Převažující interakcí během 1,3-dipolárních cykloadicí je interakce mezi největším HOMO a největším LUMO. Regioselektivita je tak řízena atomy s největšími HOMO a LUMO orbitalovými koeficienty.[22][23]
Jako příklad lze uvést cykloadici diatzomethanu na tři různé dipolarofily: methylakrylát, styren a methylcinnamát. Uhlíkový atom diazomethanu má největší HOMO, zatímco koncové alkenové uhlíky methylakrylátu a styrenu mají největší LUMO; cykloadice tedy probíhá regioselektivně na pozici C3. U methylcinnamátu oba substituenty (fenylová a methoxykarbonylová skupina) „soutěží“ v odtahování elektronů. Převládne vliv karboxylu, díky čemuž je nejelektrofilnější β uhlík. Karboxylová skupina se tak regioselektivně připojuje na pozici C3 a fenylová skupina na pozici C4.

Sterické efekty
Sterické efekty mohou výše zmíněné elektronové jevy podporovat, ale mohou též působit proti nim. V některých případech sterické efekty zcela převáží nad elektronovými a vzniká tak výhradně opačný regioizomer;[24] například diazomethan většinou reakcí s methylakrylátem vytváří 3-karboxylpyrazolin. Při zesilování sterických jevů ovšem začíná vznikat rovněž izomerní 4-karboxylpyrazolin, poměr množství těchto izomerů závisí na intenzitě sterických jevů. Zvětšení velikosti substituentů od atomu vodíku k terc-butylu zcela obrátí regioselektivitu ze 100 % 3-karboxylové na 100 % 4-karboxylové substituce.[25][26]

Využití v syntéze
1,3-dipolární cykloadice jsou významnými postupy syntézy řady důležitých pětičlenných heterocyklů jako jsou například triazoly, furany, isoxazoly a pyrrolidiny. Některé cykloadukty mohou být štěpeny za vzniku lineárního řetězce, což poskytuje další možný způsob přípravy alifatických sloučenin. Tyto reakce jsou velmi významné rovněž díky své stereospecifitě, diastereoselektivitě a regioselektivitě.
Reakce s nitriloxidy
1,3-dipolární cykloadice s nitriloxidy je často používanou maskovanou aldolovou reakcí. Cykloadiční reakcí nitriloxidu s alkenem vzniká cyklický isoxazolinový produkt, zatímco reakce s alkynem poskytuje isoxazol. Isoxazoliny i isoxazoly mohou být rozštěpeny hydrogenací za vzniku β-hydroxykarbonylových sloučenin aldolového typu nebo β-dikarbonylových sloučenin Claisenova typu.
Nitriloxidová-alkynová cykloadice následovaná hydrogenací byla využita při syntéze miyakolidu, jak je uvedeno na následujícím obrázku:[27]

Karbonylylidy
1,3-dipolární cykloadiční reakce byly vyvinuty jako výborné metody k přípravě komplexních cyklických řetězců a molekul pro lékařské, biologické a mechanistické výzkumy. Konkrétně [3+2] cykloadice se zahrnutím karbonylylidů byly široce využity na přípravu molekul s pětičlennými cykly obsahujícími kyslík.[28]
Příprava karbonylylidů pro 1,3-dipolární cykloadice
Ylidy jsou považovány za kladně nabité heteroatomy spojené se záporně nabitými uhlíkovými atomy, patří k nim též ylidy sulfonia, thiokarbonylu, oxonia, dusíku a karbonylu.[29] Karbonylylidy, důležité meziprodukty při přípravě pětičlenných cyklů obsahujících kyslík, lze připravtit několika způsoby.
Příprava karbonylylidů z derivátů diazomethanu fotokatalýzou
Jedna z prvních metod, z roku 1983, syntézy karbonylylidů v sobě zahrnuje fotokatalýzu.[30] Fotolýzou diazotetrakis(trifluormethyl)cyklopentadienu (DTTC) za přítomnosti tetramethylmočoviny lze připravit karbonylylid vnitromolekulární nukleofilní reakcí a následnou aromatizací zbylé DTTC.[30] Produkt byl izolován a poté analyzován rentgenovou krystalografií kvůli stabilitě získané díky aromaticitě, trifluormethylovým skupinám snižujícím a dimethylaminovým skupinám zvyšujícím elektronovou hustotu. Stabilní karbonylylidové dipóly mohou být následně využity k [3+2] cykloadičním reakcím s dipolarofily.

Další příklad syntézy karbonylylidů byl popsán roku 1986.[31] Dideuteriodiazomethan byl fotolyticky rozložen nza přítomnosti formaldehydu a vznikl dideuterioformaldehydový karbonylylid.

Příprava karbonylylidů z hydroxypyronů přesunem protonů
Karbonylylidy lze připravit kysele katalyzovanou reakcí z hydroxy-3-pyronů za nepřítomnosti kovového katalyzátoru.[32] Na začátku dochází k tautomerizaci, následuje eliminace odcházející skupiny, což vede k aromatizaci pyronového kruhu a vzniku karbonylylidu; z něj pak lze cykloadicí vytvořit oxacyklus. Tento způsob se nepoužívá tak často kvůli požadavkům na pyronové sloučeniny a také kvůli omezené využitelnosti.

5-hydroxy-4-pyrony lze též použít na přípravu karbonylylidů vnitromolekulárním přesunem vodíkového protonu.[33] Po provedení reakce může vzniklý karbonylylid reagovat s dipolarofily za vzniku kyslíkatých heterocyklů.

Příprava α halogenkarbonylylidů z dihalogenkarbenů
K tvorbě karbonylylidů lze využít i dihalogenkarbeny, přičemž se využívá schopnost těchto sloučenin snižovat elektronovou hustotu.[34][35][36] Jako zdroj dichlorkarbenů lze použít fenyl(trichlormethyl)rtuť a jako zdroj dibromkarbenů se dá použít fenyl(tribrommethyl)rtuť. Karbonylylid je možné získat reakcí dihalogenkarbenů s ketony nebo aldehydy. Při přípravě α halogenkarbonylylidů ovšem může dojít k nežádoucí ztrátě molekuly oxidu uhelnatého a následnému vzniku deoxygenovaného produktu.

Příprava karbonylylidů z derivátů diazomethanu za katalýzy sloučeninami kovů
Jedním z možných způsobů přípravy karbonylylidů jsou sloučeninami kovů katalyzované reakce α diazokarbonylových sloučenin, jako katalyzátor se nejčastěji používají mědné nebo rhodné sloučeniny.[37] Po uvolnění plynného dusíku a přeměně na metalokarben dochází ke vnitromolekulární reakci, jejímž produktem je karbonylylid; tetn poté cykloadiční reakcí s alkenovým nebo alkynovým dipolarofilem vytvoří pětičlenný cyklus obsahující kyslíkový atom. K nejčastějším katalyzátorům takovýchto reakcí patří Rh2(OAc)4 a Cu(acac)2.[38][39]

Azomethinylidy
1,3-dipolární cykloadiční reakcí azomethinylidu s alkenem vzniká sloučenina s azacyklickou strukturou jako například pyrrolidin. Tento postup se mimo jiné využívá při syntéze spirotryprostatinu A.[40]
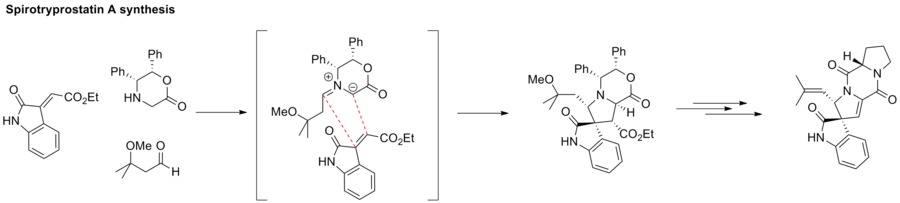
Biologická využití
1,3-dipolární cykloadice mezi organickými azidy a koncovými alkyny (například azido-alkynová Huisgenova cykloadice) se využívá v biokonjugaci.
Katalýza měďnými sloučeninami
Huisgenova reakce obecně při nízkých teplotách neprobíhá příliš rychle. Byla vyvinuta její varianta katalyzovaná měďnými sloučeninami Cu+, která probíhá velmi rychle i za mírných, například fyziologických, podmínek (neutrální pH, pokojová teplota a vodný roztok).[41][42] Tato reakce je také bioortogonální: azidy i alkyny se v biologických systémech obvykle nevyskytují a tak mohou být provedeny chemoselektivně i v rámci buňky. Tyto látky rovněž nereagují s ostatními funkčními skupinami vyskytujícími se v přírodě, a tak biologické systémy nenarušují. Měďné sloučeniny jsou cytotoxické, ovšem bylo vyvinuto mnoho ligandů, které snižují cytotoxicitu a zvyšují rychlost reakce, což umožňuje její využití při in vivo studiích.[43]

Bylo například popsáno metabolické zařazení do glykanů v buněčných membránách u azidy funkcionalizovaných sacharidů a jejich následné značkování komplexy fluoroforů s alkyny. Fluorescenčně značkovaná buněčná membrána může být zobrazena pomocí fluorescenčního mikroskopu.[44]

Napětím podporovaná cykloadice
Aby zamezili toxickým účinkům jednomocné mědi, vyvinuli Carolyn Bertozzi et al. napětím podporovanou azido-alkynovou cykloadiční reakci (SPAAC) mezi organickým azidem a derivátem cyklooktynu. Úhlové pokřivení cyklooktynu umožňuje urychlit reakci, díky čemuž ji lze prakticky provést za fyziologických podmínek i bez použití katalyzátoru.[45]

Alice Ting et al. zabudovali azidofunkcionalitu do určitých bílkovin na povrchu buněk za použití ligázového enzymu. Tyto bílkoviny byly následně označkovány cyklooktyno-fluoroforovým komplexem za vzniku fluorescenčně zbarveného proteinu.[46]

Reference
V tomto článku byl použit překlad textu z článku 1,3-Dipolar cycloaddition na anglické Wikipedii.
- ↑ HUISGEN, Rolf. 1.3-Dipolare Cycloadditionen Ruckschau und Ausblick.. Angewandte Chemie. 1963, roč. 75, s. 604–637. Dostupné online [abstract]. (anglicky)
- ↑ a b HUISGEN, Rolf. Kinetics and Mechanism of 1,3-Dipolar Cycloadditions. Angewandte Chemie International Edition. November 1963, roč. 2, čís. 11, s. 633–645. Dostupné v archivu pořízeném dne 2012-12-11. DOI 10.1002/anie.196306331. (anglicky)
- ↑ FIRESTONE, R. Mechanism of 1,3-dipolar cycloadditions. Journal of Organic Chemistry. 1968, roč. 33, s. 2285–2290. Dostupné online. DOI 10.1021/jo01270a023. (anglicky)
- ↑ HUISGEN, Rolf. 1,3-Dipolar cycloadditions. 76. Concerted nature of 1,3-dipolar cycloadditions and the question of diradical intermediates. Journal of Organic Chemistry. 1976, roč. 41, s. 403–419. Dostupné online. DOI 10.1021/jo00865a001. (anglicky)
- ↑ MLOSTON, G.; LANGHALS, E.; HUISGEN, Rolf. First Two-Step 1,2-Dipolar Cycloadditons: Nonstereospecificity. J. Am. Chem. Soc.. 1986, roč. 108, s. 6401–66402. Dostupné online. DOI 10.1021/ja00280a053. (anglicky)
- ↑ SEYYED AMIR, Siadati. An example of a stepwise mechanism for the catalyst-free 1,3-dipolar cycloaddition between a nitrile oxide and an electron rich alkene. Tetrahedron Letters. 2015, roč. 56, s. 4857–4863. Dostupné online. DOI 10.1016/j.tetlet.2015.06.048. (anglicky)
- ↑ HUISGEN, Rolf. 1,3-Dipolar Cycloadditions. Past and Future. Angewandte Chemie International Edition. 1963, roč. 2, s. 565–598. Dostupné online. DOI 10.1002/anie.196305651. (anglicky)
- ↑ COX, A; THOMAS, L; SHERIDAN, J. Microwave Spectra of Diazomethane and its Deutero Derivatives. Nature. 1958, roč. 181, čís. 4614, s. 1000–1001. Dostupné online. DOI 10.1038/1811000a0. Bibcode 1958Natur.181.1000C. (anglicky)
- ↑ HILBERTY, P; LEFORESTIER, C. Expansion of molecular orbital wave functions into valence bond wave functions. A simplified procedure.. Journal of the American Chemical Society. 1978, roč. 100, s. 2012–2017. Dostupné online. DOI 10.1021/ja00475a007. (anglicky)
- ↑ MCGARRITY, J.F.; PATAI, Saul. Basicity, acidity and hydrogen bonding. Diazonium and Diazo Groups. 1978, roč. 1, s. 179–230. Dostupné online. DOI 10.1002/9780470771549.ch6. (anglicky)
- ↑ BERNER, Daniel; MCGARRITY, John. Direct observation of the methyldiazonium ion in fluorosulfuric acid. Journal of the American Chemical Society. 1979, roč. 101, s. 3135–3136. Dostupné online. DOI 10.1021/ja00505a059. (anglicky)
- ↑ MULLER, Eugen; RUNDEL, Wolfgans. Untersuchungen an Diazomethanen, VI. Mitteil.: Umsetzung von Diazoäthan mit Methyllithium. Chemische Berichte. 1956, roč. 89, s. 1065–1071. Dostupné online. DOI 10.1002/cber.19560890436. (anglicky)
- ↑ BIHLMAIER, Werner; GEITTNER, Jochen; HUISGEN, Rolf; REISSIGP, Hans-Ulrich. The Stereospecificity of Diazomethane Cycloadditions. Heterocycles. 1978, roč. 10, s. 147–152. Dostupné online. DOI 10.3987/S-1978-01-0147. (anglicky)
- ↑ HUISGEN, Rolf; SCHEER, Wolfgang; HUBER, Helmut. Stereospecific Conversion of cis-trans Isomeric Aziridines to Open-Chain Azomethine Ylides. Journal of the American Chemical Society. 1967, roč. 89, s. 1753–1755. Dostupné online. DOI 10.1021/ja00983a052. (anglicky)
- ↑ DAHMEN, Alexander; HAMBERGER, Helmut; HUISGEN, Rolf; MARKOWSKI, Volker. Conrotatory ring opening of cyanostilbene oxides to carbonyl ylides. Journal of the Chemical Society D: Chemical Communications. 1971, čís. 19, s. 1192–1194. Dostupné online. DOI 10.1039/C29710001192. (anglicky)
- ↑ PADWA, Albert; SMOLANOFF, Joel. Photocycloaddition of arylazirenes with electron-deficient olefins. Journal of the American Chemical Society. 1971, roč. 93, s. 548–550. Dostupné online. DOI 10.1021/ja00731a056. (anglicky)
- ↑ IWASHITA, Takashi; KUSUMI, Takenori; KAKISAWA, Hiroshi. A Synthesis of dl-isoretronecanol. Chemistry Letters. 1979, čís. 11, s. 1337–1340. Dostupné online. DOI 10.1246/cl.1979.1337. (anglicky)
- ↑ WANG, Chia-Lin; RIPKA, William; CONFALONE, Pat. A short and stereospecific synthesis of (±)-α-lycorane. Tetrahedron Letters. 1984, roč. 25, s. 4613–4616. Dostupné online. DOI 10.1016/S0040-4039(01)91213-4. (anglicky)
- ↑ KANEMASA, Shuji. Metal-Assisted Stereocontrol of 1,3-Dipolar Cycloaddition Reactions. Synthesis Letters. 2002, roč. 2002, s. 1371–1387. Dostupné online. DOI 10.1055/s-2002-33506. (anglicky)
- ↑ BODE, Jeffrey; CARREIRA, Erick. Stereoselective Syntheses of Epothilones A and B via Directed Nitrile Oxide Cycloaddition.. Journal of the American Chemical Society. 2011, roč. 123, čís. 15, s. 3611–3612. Dostupné online. DOI 10.1021/ja0155635. PMID 11472140. (anglicky)
- ↑ Vsevolod V. Rostovtsev; Luke G. Green; Valery V. Fokin; K. Barry Sharpless. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective Ligation of Azides and Terminal Alkynes. Angewandte Chemie International Edition. 2002, roč. 41, čís. 14, s. 2596–22599. DOI 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. PMID 12203546. (anglicky)
- ↑ CARAMELLA, Pierluigi; HOUK, K.N. Geometries of nitrilium betaines. The clarification of apparently anomalous reactions of 1,3-dipoles. Journal of the American Chemical Society. 1976, roč. 98, s. 6397–6399. Dostupné online. DOI 10.1021/ja00436a062. (anglicky)
- ↑ CARAMELLA, Pierluigi; GANDOUR, Ruth W.; HALL, Janet A.; DEVILLE, Cynthia G. A derivation of the shapes and energies of the molecular orbitals of 1,3-dipoles. Geometry optimizations of these species by MINDO/2 and MINDO/3. Journal of the American Chemical Society. 1977, roč. 99, s. 385–392. Dostupné online. DOI 10.1021/ja00444a013. (anglicky)
- ↑ HUISGEN, Rolf. Kinetics and Mechanism of 1,3-Dipolar Cycloadditions. Angewandte Chemie International Edition. November 1963, roč. 2, čís. 11, s. 633–645. Dostupné v archivu pořízeném dne 2012-12-11. DOI 10.1002/anie.196306331. (anglicky)
- ↑ PADWA, Albert. 1,3-Dipolar Cycloaddition Chemistry. United States of America: Wiley-Interscience, 1983. ISBN 0-471-08364-X. S. 141–145.
- ↑ KOSZINOWSKI, J.. thesis. 1980. Ph.D..
- ↑ EVANS, David; RIPIN, David; HALSTEAD, David; CAMPOS, Kevin. Synthesis and Absolute Stereochemical Assignment of (+)-Miyakolide. Journal of the American Chemical Society. 1999, roč. 121, s. 6816–6826. Dostupné online. DOI 10.1021/ja990789h. (anglicky)
- ↑ Synthetic Reactions of M=C and M=N Bonds: Ylide Formation, Rearrangement, and 1,3-Dipolar Cycloaddition; Hiyama, T. W., J., Ed.; Elsevier, 2007; Vol. 11.
- ↑ Padwa, A.; Hornbuckle, S. F. Ylide Formation from the Reaction of Carbenes and Carbenoids with Heteroatom Lone Pairs Chem Rev 1991, 91, 263.
- ↑ a b Janulis, E. P.; Arduengo, A. J. Structure of an Electronically Stabilized Carbonyl Ylide J Am Chem Soc 1983, 105, 5929.
- ↑ Prakash, G. K. S.; Ellis, R. W.; Felberg, J. D.; Olah, G. A. Formaldehyde 0-Methylide, [CH2=O+-CH2: The Parent Carbonyl Ylide] J Am Chem Soc 1986, 108, 1341.
- ↑ Sammes, P. G.; Street, L. J. Intra molecular Cyclo additions with Oxido pyrylium Ylides J. Chem. Soc., Chem. Commun. 1982, 1056.
- ↑ Garst, M. E.; McBride, B. J.; Douglass III, J. G. Intramolecular cycloadditions with 2-(ω-alkenyl)-5-hydroxy-4-pyrones Tetrahedron Lett. 1983, 24, 1675.
- ↑ Gisch, J. F.; Landgrebe, J. A. Dichlorocarbene from flash vacuum pyrolysis of trimethyl(trichloromethyl)silane. Possible observation of 1,1-dichloro-3-phenylcarbonyl ylide J Org Chem 1985, 50, 2050.
- ↑ Huan, Z. W.; Landgrebe, J. A.; Peterson, K. Dibromocarbonyl ylides: Deoxygenation of aldehydes and ketones by dibromocarbene J Org Chem 1983, 48, 4519.
- ↑ Martin, C. W.; Lund, P. R.; Rapp, E.; Landgrebe, J. A. Halogenated carbonyl ylides in the reactions of mercurial dihalocarbene precursors with substituted benzaldehydes J Org Chem 1978, 43, 1071.
- ↑ Hodgson, D. M.; Bruckl, T.; Glen, R.; Labande, A. H.; Selden, D. A.; Dossetter, A. G.; Redgrave, A. J. Catalytic enantioselective intermolecular cycloadditions of 2-diazo-3,6-diketoester-derived carbonyl ylides with alkene dipolarophiles Proceedings of the National Academy of Sciences of the United States of America 2004, 101, 5450.
- ↑ Padwa, A.; Hertzog, D. L.; Nadler, W. R. Intramolecular Cycloaddition of Isomunchnone Dipoles to Heteroaromatic π-Systems J Org Chem 1994, 59, 7072.
- ↑ Hamaguchi, M.; Ibata, T. New Type of Mesoionic System. 1,3-Dipolar Cycloaddition of Isomunchnon With Ethylenic Compounds Chem Lett 1975, 499.
- ↑ ONISHI, Tomoyuki; SEBAHAR, Paul; WILLIAMS, Robert. Concise, Asymmetric Total Synthesis of Spirotryprostatin A. Organic Letters. 2003, roč. 5, s. 3135–3137. Dostupné online. DOI 10.1021/ol0351910. PMID 12917000. (anglicky)
- ↑ TORNOE, Christian; CHRISTENSEN, Caspar; MELDAL, Morten. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. Journal of Organic Chemistry. 2002, roč. 67, čís. 9, s. 3057–3064. DOI 10.1021/jo011148j. PMID 11975567. (anglicky)
- ↑ ROSTOVTSEV, Vsevolod; GREEN, Luke; FOKIN, Valery; SHARPLESS, Barry K. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective Ligation of Azides and Terminal Alkynes. Angewandte Chemie International Edition. 2002, roč. 41, s. 2596–2599. DOI 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. PMID 12203546. (anglicky)
- ↑ BESANCENEY-WEBLER, Christen; JIANG, Hao; ZHENG, Tianqing; FENG, Lei. Increasing the Efficacy of Bioorthogonal Click Reactions for Bioconjugation: A Comparative Study. Angewandte Chemie International Edition. 2011, roč. 50, s. 8051–8056. Dostupné online. DOI 10.1002/anie.201101817. PMID 21761519. (anglicky)
- ↑ BREIDENBACH, Mark; GALLAGHER, Jennifer; KING, David; SMART, Brian. Targeted metabolic labeling of yeast N-glycans with unnatural sugars. Proceedings of the National Academy of Sciences of the United States of America. 2010, roč. 107, čís. 9, s. 3988–3993. Dostupné online. DOI 10.1073/pnas.0911247107. PMID 20142501. Bibcode 2010PNAS..107.3988B. (anglicky)
- ↑ AGARD, Nicholas; PRESCHER, Jennifer; BERTOZZI, Carolyn. A Strain-Promoted [3 + 2] Azide−Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. Journal of the American Chemical Society. 2004, roč. 126, s. 15046–15047. DOI 10.1021/ja044996f. PMID 15547999. (anglicky)
- ↑ FERNANDEZ-SUAREZ, Marta; BARUAH, Hemanta; MARTINEZ-HERNANDEZ, Laura; XIE, Kathleen. Redirecting lipoic acid ligase for cell surface protein labeling with small-molecule probes. Nature Biotechnology. 2007, roč. 25, s. 1483–1487. DOI 10.1038/nbt1355. PMID 18059260. (anglicky)
Externí odkazy
 Obrázky, zvuky či videa k tématu 1,3-dipolární cykloadice na Wikimedia Commons
Obrázky, zvuky či videa k tématu 1,3-dipolární cykloadice na Wikimedia Commons