
Metalace
Metalace jsou chemické reakce, při kterých se navazují atomy kovu na organické sloučeniny, obvykle přitom nahrazují atomy halogenů. V laboratořích se obvykle používají k aktivaci organických molekul v průběhu vytváření vazeb C—X (kde X je většinou uhlík, kyslík nebo dusík), jež jsou potřebné k tvorbě dalších organických sloučenin.
Metalovaná činidla mívají nejčastěji využití při nukleofilních substitucích, reakcích s přenosem jednoho elektronu a redoxních reakcích funkčních skupin na jiných molekulách (jako jsou ketony, aldehydy a halogenalkany). Metalované molekuly se také účastní acidobazických reakcí, kde organokovové činidlo deprotonuje organickou molekulu za vzniku nového organokovového činidla.
Nejrozšířenější skupinou metalovaných sloučenin jsou organolithné sloučeniny a Grignardova činidla; v laboratořích i průmyslu se však používají i jiné organokovové sloučeniny, například organozinečnaté sloučeniny.
Historie
[editovat | editovat zdroj]Metalaci v laboratoři poprvé pozoroval Edward Frankland při syntéze diethylzinku v roce 1849.[1]
Tento objev vedl k vývoji organokovových sloučenin odvozených od jiných kovů,[2] které ovšem měly jen omezené využití v laboratořích, protože byly drahé a značně pyroforické. Rozvoj metalací (převážně transmetalací) nastal až po Grignardovu objevu přípravy organohořečnatých halogenidů přímo z kovového hořčíku a organohalogenidů.[3]
Tato organohořečnatá činidla našla díky možnosti provádět metalace mnoha různých substrátů široké využití v laboratořích.[4]
Organolithné sloučeniny byly poprvé připraveny roku 1917,[5] ovšem jejich využívání se rozšířilo až poté, kdy Karl Ziegler, Henry Gilman a Georg Wittig vyvinuly jednodušší metody jejich přípravy.[6]
Po objevu těchto vylepšení došlo k výraznému nárůstu zájmu o tyto sloučeniny, protože jsou většinou reaktivnější než organohořečnatá činidla. První použití organolithného činidla k metalaci bylo popsáno v roce 1928, šlo o reakci fluorenu s ethyllithiem.[7]
Reaktivita a použití
[editovat | editovat zdroj]Mnoho jednou metalovaných sloučenin lze zakoupit v pevném skupenství i jako roztoky, přičemž roztoky jsou dostupné v řadě různých rozpouštědel i koncentrací. Také je lze připravit v laboratoři jako in situ meziprodukty nebo jako roztoky.
Reaktivita metalovaných sloučenin
[editovat | editovat zdroj]Velký rozdíl v elektronegativitě atomů uhlíku a kovu u většiny metalovaných sloučenin způsobuje značnou polaritu vazby uhlík-kov. Vysoká polarita vazby a z toho vyplývající vysoká elektronová hustota okolo metalovaného uhlíkového atomu způsobuje, že se povaha vazby blíží vazbě iontové. Metalované sloučeniny tak bývají dobrými nukleofily a silnými zásadami.
Metalované sloučeniny mají největší využití v organické syntéze, kde slouží jako nukleofily při nukleofilních substitucích, jako silné zásady při deprotonacích, iniciátory polymerizací a jako výchozí materiály pro přípravu dalších organokovových sloučenin transmetalačními reakcemi.
Stericky narušované metalované sloučeniny, například komplexy n-butyllithia se často používají jako superzásady či iniciátory polymerizací, protože jimi vyvolávané sterické efekty narušují možnosti sloučenin přiblížit se k nukleofilům dostatečně blízko na provedení nukleofilního ataku. Sloučeniny bez těchto vlastností, například methyllithium a alkylhořečnaté halogenidy, se většinou používají jako nukleofily nebo transmetalační činidla, i když jejich silně zásaditá povaha často vyžaduje ochranu zásaditých funkčních skupin nacházejících se v organických molekulách.
Mechanismus
[editovat | editovat zdroj]Metalace se obvykle využívá k syntéze složitějších organokovových sloučenin, jako jsou alkynyllithná činidla, z uhlovodíků obsahujících kyselé vodíky. Při mezimolekulárních i vnitromolekulárních metalacích reakce probíhá přes acidobazickou funkcionalizaci vazby C-H párováním kovu (M) se zásadou (B) podle následujícího schématu:

Relativní stabilita konečných produktů závisí na tom, zda je reakce vratná či nevratná a relativní kyselost vazeb C-H v metalovaných molekulách určuje, kam v molekule reaktantu se kov naváže.
První mechanismus metalace, založený na soustředěné metalaci-deprotonaci (CMD), navrhli v roce 1955 S. Winstein a T. G. Traylor na základě elektrofility rtuti při acetolýze difenylrtuti v kyselině octové.[8]
Pozdější mechanistické studie podpořily tento mechanismus u mezimolekulárních i vnitromolekulárních metalací. Všeobecně přijímaný mechanismus je znázorněn níže, jako příklad je zde použita metalace primárního vodíku.[9]
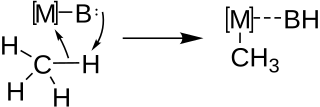
Mechanismus kovem a zásadou řízené funkcionalizace vazby C-H na primárním vodíku

Transmetalace
[editovat | editovat zdroj]Transmetalace je výměna dvou kovů mezi organickými molekulami prostřednictvím redoxní záměny. Často jde o reakci organolithného činidla se solí kovu.
Organolithné sloučeniny
[editovat | editovat zdroj]Při přípravě jednoduchých organolithných činidel redukcí jednoho ekvivalentu alkyl- nebo arylhalogenidu dvěma ekvivalenty lithia vzniká jeden ekvivalent alkyl- nebo aryllithné sloučeniny a jeden ekvivalent lithného halogenidu.[10]

Reakce probíhá radikálovým mechanismem a pravděpodobně je iniciována jednoelektronovým mechanismem (znázorněným níže).[11]

Podobně může hořčík metalovat organohalogenidy za vzniku Grignardových činidel
Reference
[editovat | editovat zdroj]V tomto článku byl použit překlad textu z článku Metalation na anglické Wikipedii.
- ↑ E. Frankland. Ueber die Isolirung der organischen Radicale. European Journal of Organic Chemistry. 1849, s. 171–213. Dostupné online. DOI 10.1002/jlac.18490710205.
- ↑ Johnson, W.C. Die Chemie der Metall-Organischen Verbindungen (Krause, Erich; Grosse, A. V.). Journal of Chemical Education. 1939, s. 148. DOI 10.1021/ed016p148.1. Bibcode 1939JChEd..16..148J.
- ↑ V. Grignard. Sur quelques nouvelles combinaisons organométaliques du magnésium et leur application à des synthèses d'alcools et d'hydrocabures. Comptes rendus de l'Académie des Sciences. 1900, s. 1322–1325. Dostupné online.
- ↑ John J. Eisch. Henry Gilman: American Pioneer in the Rise of Organometallic Chemistry in Modern Science and Technology. Organometallics. 2002, s. 5439–5463. DOI 10.1021/om0109408.
- ↑ W. Schlenk; J. Holtz. Über die einfachsten metallorganischen Alkaliverbindungen. European Journal of Inorganic Chemistry. 1917, s. 262–274. Dostupné online. DOI 10.1002/cber.19170500142.
- ↑ H. Gilman; E. A. Zoellner; W. M. Selby. An Improved Procedure for the Preparation of Organolithium Compounds. Journal of the American Chemical Society. 1932, s. 1957–1962. DOI 10.1021/ja01344a033.
- ↑ Schlenk, Bergmann. II. Neuartige Erkenntnisse auf dem Gebiete der Stereochemie des Kohlenstoffs. Justus Liebig's Annalen der Chemie. 1928, s. 192. DOI 10.1002/jlac.19284630103.
- ↑ S. Winstein; T. G. Traylor. Mechanisms of Reaction of Organomercurials. II. Electrophilic Substitution on Saturated Carbon. Acetolysis of Dialkylmercury Compounds. Journal of the American Chemical Society. 1955, s. 3747–3752. DOI 10.1021/ja01619a021.
- ↑ D. Lapointe; K. Fagnou. Overview of the Mechanistic Work on the Concerted Metallation–Deprotonation Pathway. Chemistry Letters. 2010, s. 1118–1126. Dostupné online. DOI 10.1246/cl.2010.1118.
- ↑ Organometallics in Organic Synthesis, Schlosser, M., Ed, Wiley: New York, 1994. ISBN 0-471-93637-5
- ↑ William F. Bailey; Jeffrey J. Patricia. The mechanism of the lithium - halogen Interchange reaction : a review of the literature. Journal of Organometallic Chemistry. 1988, s. 1–46. DOI 10.1016/0022-328x(88)83017-1.