
Borylace
Borylace jsou skupina organických reakcí katalyzovaných přechodnými kovy, kterými vznikají organoborité sloučeniny funkcionalizací alifatických a aromatických vazeb C–H; díky tomu mají význam pro aktivace vazeb uhlík-vodík.[1] Kovy katalyzované borylace vazeb C–H umožňují pomocí přechodných kovů k přímé přeměně vazeb C–H na vazby C–B. Tento postup může být výhodný oproti tradičním borylacím, protože umožňuje použití cenově dostupných uhlovodíkových prekurzorů, omezení využívání prefunkcionalizovaných organických sloučenin, toxických vedlejších produktů a usnadnění syntézy biologicky významných molekul.[2][3] Častými borylovými skupinami zaváděnými do organických sloučenin borylačními reakcemi jsou boronové kyseliny a jejich estery.[4] Boronové kyseliny jsou trojvazné organické sloučeniny obsahující bor, na který jsou navázány jedna alkylová nebo arylová a dvě hydroxylové skupiny, jejich estery mají jeden alkylový nebo arylový a dva esterové substituenty. Boronové kyseliny a estery se dělí na základě druhu uhlovodíkové skupiny (R) navázané přímo na bor, například na alkyl-, alkenyl-, alkynyl- and arylboronové estery. Nejběžnější typ boronových esterů využívaných k borylacím organických sloučenin katalyzovaných přechodnými kovy má obecný vzorec (RO)2B-B(OR)2; patří sem například bis(pinakoláto)dibor (B2Pin2) a bis(katecholáto)diboran (B2Cat2).[5]

Atomy boru v boronových kyselinách a esterech jsou sp2 hybridizované s prázdnými p orbitaly, které jim dodávají vlastnosti odpovídající Lewisovým kyselinám. Vazby C–B u boronových kyselin a esterů jsou poněkud delší než běžné vazby C–C, s obvyklými délkami 155-159 pm. Prodloužení vazeb C–B oproti C–C vede k tomu, že tyto vazby mají i o něco nižší energie než vazby C-C (323 kJ/mol u C–B a 358 kJ/mol u C–C).[6] Vazby mezi uhlíkovými a vodíkovými atomy vykazují délky kolem 109 pm a energie přibližně 413 kJ/mol. Vazby C–B jsou tak vhodnými meziprodukty nahrazujícími obvykle nereaktivní vazby C–H.
Sloučeniny obsahující vazby mezi atomy uhlíku a boru se nazývají organoborité sloučeninyy. Mají široké využití v syntetických reakcích, protože se vazby C–B bond dají snadno přeměnit na vazby C–X (X = Br, Cl), C–O, C–N nebo C–C bond. Vzhledem k užitečnosti vazeb C–B bylo vyvinuto mnoho různých postupů na jejich zavádění do organických sloučenin.[7]
Organoborité sloučeniny se často připravují z Grignardových činidel hydroboračními nebo diboračními reakcemi.[8]
Kovy katalyzované C–H borylace[editovat | editovat zdroj]
Alifatické C–H borylace[editovat | editovat zdroj]
Alkany mohou být selektivně borylovány na primárních vazbách C–H s využitím Cp*Rh(η4-C6Me6) jako katalyzátoru.[9]
Vysoká selektivita na primárních vazbách C–H se objevuje i tehdy, když jsou v hlavních řetězcích molekul substrátu přítomny heteroatomy. Rhodiem katalyzované borylace vazeb C–H v methylových skupinách jsou selektivní nezávisle na poloze heteroatomu. K borylaci dochází na nejméně stericky zatížené a na elektrony nejchudší primární vazbě C–H acetalů, etherů, aminů a alkylfluoridů.[10]
Při nepřítomnosti primárních vazeb C–H v molekule substrátu, například u cyklohexanu, k reakci nedochází.

Selektivita funkcionalizací primárních alkanových vazeb je způsobena tvorbou kineticky a termodynamicky výhodných primárních alkylkovových komplexů přes sekundární alkylkovové komplexy.[11]

Větší stabilita primárního alkylkomplexu oproti sekundárnímu je dána několika faktory; Prvním je jeho sterická výhodnost, druhým to, že se na α-uhlících alkylkovových komplexů často nacházejí částečné záporné náboje a primární alkylové ligandy si drží částečné záporné náboje lépe než sekundární ligandy. Příčiny selektivity C–H borylací pomocí katalyzátorů založených na rhodiu byla zkoumána pomocí druhu mechanistické studie nazývané vodíko-deuteriová výměna. Ukázalo se, že regioselektivita procesu je způsobována přednostním štěpením primárních vazeb C–H a funkcionalizací primárního alkylkovového meziproduktu oproti sekundárnímu.[12]

Syntetické možnosti borylací vazeb C–H bylo využito k modifikacím polymerů borylacemi a následnými oxidacemi za vzniku hydroxylových polymerů.[13]

Aromatické C–H borylace[editovat | editovat zdroj]
Stericky řízené C–H borylace arenů[editovat | editovat zdroj]
První případ katalytické borylace vazby C–H u neaktivovaného uhlovodíku (konkrétně benzenu) popsali Milton R. Smith a Carl N. Iverson; jako katalyzátor byl použit komplex Ir(Cp*)(H)(Bpin). Účinnost byla ovšem nízká, když se za 120 h při 150 °C podařilo provést pouze tři cykly.[14]
Později bylo vyvinuto několik vylepšení, které vedly k účinným borylacím arenů za vhodných podmínek. Aromatické C–H borylace rozvinuli John F. Hartwig a Išijama pomocí bis(pinacoláto)dibou, katalyzátory byly 4,4’-di-terc-butylbipyridin (dtbpy) a bis[(cyklooktadien]di-μ-methoxydiiridium.[15]
S tímto katalytickým systémem probíhaly borylace arenů s regioselektivitou řízenou stetrickými efekty použitých arenů. Selektivitu funkcionalizací aromatických vazeb C–H ovlivňuje pravidlo, že reakce neprobíhá v pozici ortho vůči substituentu, jestliže je dostupná vazba C–H, která nemá ortho substituent.[11] Pokud je přítomna pouze jedna funkční skupina, tak borylace probíhá do poloh meta a para v poměru 2:1. Ortho izomer nevzniká kvůli sterickým efektům vytvářeným substituentem.[16]

Adice Bpin probíhá u symetricky 1,2- a 1,4-substituovaných arenů pouze do jedné pozice. Symetricky i nesymetricky 1,3-substituované areny se také borylují selektivně, protože je stericky dostupná jen jedna vazba C–H.

Tímto se reakce liší od elektrofilních aromatických substitucí, kde regioselektivitu řídí elektronové efekty.[17]

Níže je zobrazeno syntetické využití C–H borylace arenů, kdy je možné převést 1,3-disubstituované aromatické sloučeniny přímo na 1,3,5-organoborité molekuly a následně je funkcionalizovat.[15]

Aromatické funkcionalizace vazeb C–H byly využity při totální syntéze plavuňového alkaloidu komplanadinu A, posilujícího expresi mRNA nervového růstového faktoru a jeho tvorbu v lidských neurogliových buňkách. Přírodní látky podporující růst nových nervových sítí jsou předmětem zájmu co se týče léčby nemocí, jako je Alzheimerova choroba.[18] Komplanadin A byl připraven kombinací přímé aromatické C–H borylace a Suzukiovy reakce, po které následovalo odštěpení terc-butyloxykarbonylové chránicí skupiny.

C–H borylace heteroarenů[editovat | editovat zdroj]
I heteroareny mohou být borylovány s využitím iridiových katalyzátorů, ovšem elektronové efekty zde způsobují vedlejší selektivitu, kde furany, pyrroly a thiofeny reagují na C–H vazbách v polohách alfa oproti heteroatomům. Tato selektivita je pravděpodobně způsobena tím, že jde o nejkyselejší, a tedy i nejreaktivnější, vazby C–H.[11]

Řízené ortho C–H borylace[editovat | editovat zdroj]
Stejný katalytický systém může být použit k dosažení regioselektivity bez využití substituentů, T. A. Boebel a J. F. Hartwig například provedli ortho-borylaci, při které dimethylhydrosilylové řídicí skupiny vstupují do iridiem katalyzovaných borylací vazeb C–H do polohy ortho oproti silanovým skupinám.[19] Selektivita v pozici ortho při použití hydrosilylových skupin se připisuje zpětné adici vazby Si-H na kovové centrum, jež vede k přednostnímu štěpení vazby C–H v poloze ortho vůči hydrosilylovému substituentu. Bylo vyvinuto i několik dalších postupů ortho-borylace arenů zahrnujících jiné řídicí skupiny.[20][21][22]

Mechanistické parametry C–H borylací arenů[editovat | editovat zdroj]
K usnadnění vysvětlení mechanismů C–H borylačních reakcí u arenů a heteroarenů byl použit trisboryliridiový komplex. Kineticlými studiemi studies a pomocí izotopového značkování bylo zjištěno, že s arenem v katalytickém procesu reaguje triborylový komplex iriditého kationtu.[23]
Níže je zobrazena varianta katalytického cyklu pro orthoborylaci hydrosilanů. Z kinetických dat vyplývá, že pozorovaný trisborylový komplex se koordinuje s cyklooktenem a zvratně se disociuje na šestnáctielektronový komplex. Jako řídicí skupina zde byl použit benzyldimethylsilan, o kterém se předpokládá, že reaguje s trisboryliriditým katalyzátorem skrze zvratnou adici vazby Si-H na kovové centrum, po níž následuje selektivní ortho-C–H aktivace přes oxidační adici a redukční eliminaci.[24]

Metaselektivní borylace[editovat | editovat zdroj]
Metaselektivní C–H borylace je významným syntetickým postupem, objeveným v roce 2002. Tyto reakce jsou ovšem zcela stericky řízené a mohou probíhat jen u 1,3-disubstituovaných benzenů. Přibližně o 12 let dříve byla provedena metaselektivní aktivace vazeb C–H a následná borylace. Za použití stejného substrátu je možné změnit poziční selektivitu použitím jiného ligandu. Byly navrženy dvě příčiny metaselektivity, elektrostatické interakce a sekundární interakce mezi atomy boru a dusíku.[25]
Ve stejné době skupina vědců z Japonska popsala mechanismus metaselektivní borylace založený na sekundárních interakcích. Tento mechanismus odpovídá borylacím mnoha různých karbonylových sloučenin.[26]
Redukční reakce organoboritých sloučenin[editovat | editovat zdroj]
Coreyova–Itsunova redukce[editovat | editovat zdroj]
V roce 1981 byla objevena asymetrická redukce prochirálních aromatických ketonů chirálními aminoalkoholy a boranem za vzniku příslušných sekundárních alkoholů s 60procentním enantiomerním přebytkem. Chirální aminoalkoholy reagují s boranem za tvorby aloxylaminoboranových komplexů. Komplexy obsahují poměrně nereaktivní pětičlenný kruh, který jim dodává tepelnou a hydrolytickou stabilitu a rozpustnost v řadě protických i aprotických rozpouštědel.[27]
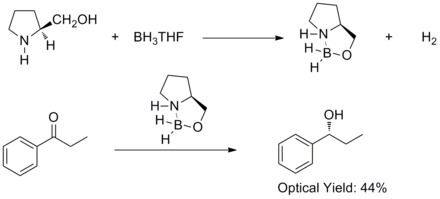
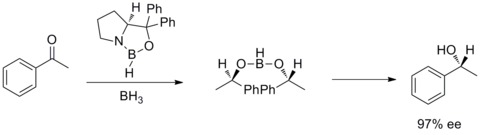
V roce 1987 Elias James Corey popsal tvorbu oxazaborolidinů z boranu a chirálních aminoalkoholů a zjistil, že oxazaborolidiny mohou katalyzovat rychlé a velmi enantioselektivní redukce prochirálních ketonů za přítomnosti BH3THF. Tato reakce bývá nazývána Coreyova–Itsunova redukce.[28][29]
Midlandova redukce[editovat | editovat zdroj]
V roce 1977 popsal M. M. Midland se svými spolupracovníky přípravu B-3-alfa-pinanyl-9-borabicyklo [3,3,1]nonanu hydroborací (+)-alfa-pinenu pomocí 9-borabicyklo[3,3,1] nonanu (9-BBN) ajeho využití k redukci benzaldehydu-alfa-d na (S)-(+)-benzyl-alfa-d-alkohol kvantitativní asymetrickou indukcí.[30]
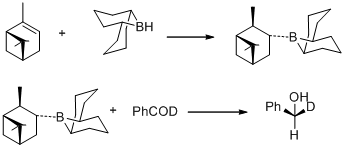
Ve stejném roce M. M. Midland zjistil, že lze jako redukční činidlo použít B-3-alfa-pinanyl-9-BBN, snadno dostupný pomocí reakce (+)-alfa-pinenu s 9-BBN.[31]
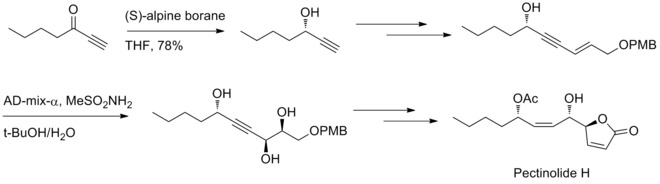
V roce 2012 oznámil U. R. Y. Venkateswarlu se spolupracovníky stereoselektivní přípravu pektinolidu zahrnující tvorbu trojice chirálních center na C–4’, C–5 a C–1’ Midlandovu redukci a Sharplessovu dihydroxylaci.[32]
Párovací reakce organoboritých sloučenin[editovat | editovat zdroj]
Petasisova reakce[editovat | editovat zdroj]
Roku 1993 oznámili Nicos A. Petasis a Irini Akritopoulou objev účinné syntézy allylaminů pomocí pozměněné Mannichovy reakce. Zjistili, že vinylboronové kyseliny se mohou jako nukleofily účastnit reakcí, při kterých vznikají geometricky čisté allylaminy. Tato upravená Mannichova reakce je nazývána Petasisova-Mannichova reakce boronových kyselin nebo, zkráceně, Petasisova reakce.[33][34]
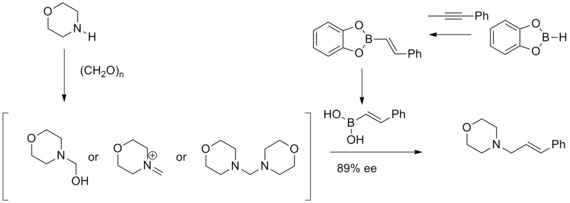
Roushova asymetrická allylace[editovat | editovat zdroj]
V roce 1978 popsali R. W. Hoffmann a T. Herold enantioselektivní syntézu sekundárních homoallylových alkoholů přes chirální neracemické allylboronové estery. Homoallylové alkoholy se tvořily s velmi dobrou výtěžností a středně velkou enantioselektivitou.[35]
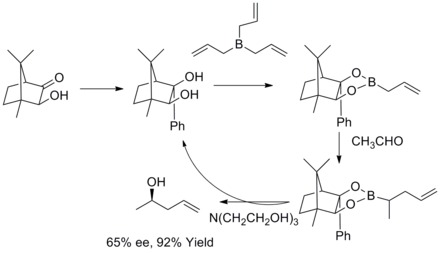
V roce 1985 W. R. Roush zjistil, že allylboronáty s navázanými tartarátovými skupinami je možné snadno použít k řízení faciální selektivity reakcí chirálních a nechirálních aldehydů. Později tento postup rozšířil na přípravu but-2-en-1,4-diolů a anti-diolů. Tato reakce je známa jako Rouchova asymetrická allylace.[36][37][38][39]
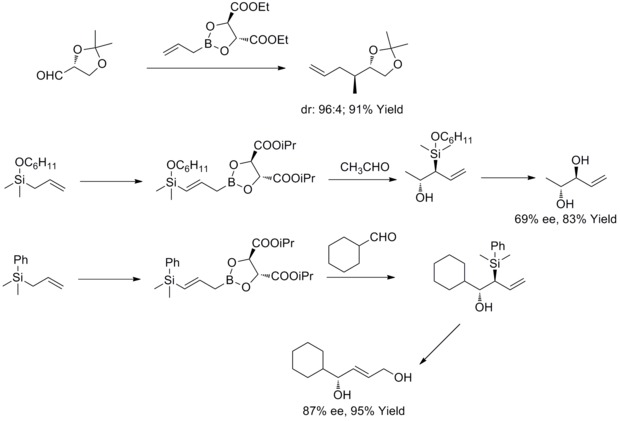
Roku 2011 provedli R. A. Fernandes a P. Kattanguru vylepšenou totální syntézu (8S, 11R, 12R)- a (8R, 11R, 12R)-diastereomerů topsentolidu B2, skládající se z osmi kroků. Jedním z kroků byla diastereoselektivní Roushova allylace.[40]

Suzukiovy reakce[editovat | editovat zdroj]
V roce 1979 oznámili N. Miyaura a A. Suzuki přípravu (E)-alkenů s vysokou výtěžností z arylhalogenidů a alkyl-1-enylboranů katalyzovanou tetrakis(trifenylfosfin)palladiem a zásadami. Poté A. Suzuki se svými spolupracovníky rozšířil tento druh reakcí nba ostatní organoborité sloučeniny a zvětšil také okruh použitelných alkenyl, aryl a alkylhalogenidů a triflátů. Palladiem katalyzované reakce organoboitých sloučenin s organohalogenidy vytvářející vazby uhlík-uhlík jsou známy jako Suzukiovy reakce.[41][42]
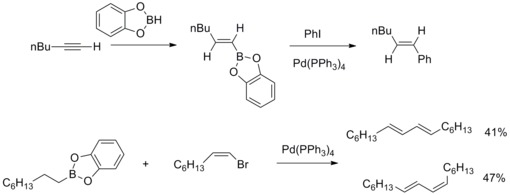
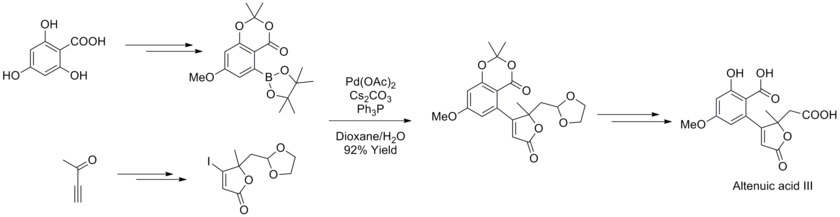
V roce 2013 určil Joachim Podlech strukturu mykotoxinu kyseliny altenuové pomocí NMR spektroskopie a provedl jeho totální syntézu. Součástí syntézy byla Suzukiova reakce, na níž byly použity vysoce funkcionalizovaný boronát a butenolidy.[43]
Pozměněná Ullmannova syntéza biaryletherů a biarylaminů[editovat | editovat zdroj]
V roce 1904 zjistil Fritz Ullmann, že použití práškové mědi výrazně usnadňuje reakce arylhalogenidů s fenoly za tvorby biaryletherů; tato reakce se nazývá Ullmannova kondenzace. Roku 1906 rozšířil možnosti Ulmannovy kondenzace na syntézu arylaminů reakcemi arylhalogenidů s amidy za přítomnosti uhličitanu draselného a jodidu měďného; tato varianta se nazývá Goldbergova modifikace Ullmannovy kondenzace.[44]
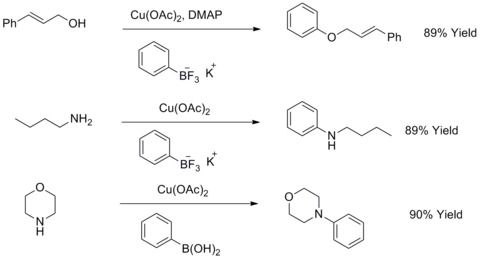
V roce 2003 popsali R. A. Batey a T. D. Quach obměnu tohoto druhu reakcí, kde reagují organotrifluorboritanové draselné soli s alifatickými alkoholy, alifatickými aminy nebo s aniliny za vzniku aryletherů či arylaminů.[45][46]
Odkazy[editovat | editovat zdroj]
Související články[editovat | editovat zdroj]
- Organoborité sloučeniny
- Coreyova–Itsunova redukce
- Midlandova redukce
- Petasisova reakce
- Suzukiova reakce
Externí odkazy[editovat | editovat zdroj]
 Obrázky, zvuky či videa k tématu Borylace na Wikimedia Commons
Obrázky, zvuky či videa k tématu Borylace na Wikimedia Commons
Reference[editovat | editovat zdroj]
V tomto článku byl použit překlad textu z článku Borylation na anglické Wikipedii.
- ↑ HARTWIG, John F. Borylation and Silylation of C–H Bonds: A Platform for Diverse C–H Bond Functionalizations. Accounts of Chemical Research. 2012, s. 864–873. ISSN 0001-4842. DOI 10.1021/ar200206a. PMID 22075137.
- ↑ J. Y. Cho; M. K. Tse; D. Holmes; R. E. Maleczka; M. R. Smith. Remarkably Selective Iridium Catalysts for the Elaboration of Aromatic C-H Bonds. Science. 2001, s. 305–308. DOI 10.1126/science.1067074. PMID 11719693.
- ↑ Ishiyama, T.; Nobuta, Y.; Hartwig, J. F.; Miyaura, N. Chem. Commun. 2003, 2924.
- ↑ Brown, H. C.; Kramer, G. W.; Levy, A. B.; Midland, M. M. Organic Synthesis via Boranes; Wiley-Interscience: New York, 1975; Vol. 1.
- ↑ H. Braunschweig; F. Guethlein. Transition-Metal-Catalyzed Synthesis of Diboranes(4). Angewandte Chemie International Edition. 2011, s. 12 613 – 12 616. DOI 10.1002/anie.201104854. PMID 22057739.
- ↑ Hall, D. G. (2011) Structure, Properties, and Preparation of Boronic Acid Derivatives, in Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials (Volume 1 and 2), Second Edition (ed D. G. Hall), Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany. DOI:10.1002/9783527639328.ch1
- ↑ Ibraheem A. I. Mkhalid; Jonathan H. Barnard; Todd B. Marder; Jaclyn M. Murphy; John F. Hartwig. C–H Activation for the Construction of C–B Bonds. Chemical Reviews. 2010, s. 890–931. DOI 10.1021/cr900206p. PMID 20028025.
- ↑ Wade, L. G., Organic Chemistry. Upper Saddle River: Pearson Education, Inc., 2010.
- ↑ H. Chen; S. Schlecht; T. C. Semple; John F. Hartwig. Thermal, Catalytic, Regiospecific Functionalization of Alkanes. Science. 2000, s. 1995–1997. DOI 10.1126/science.287.5460.1995. PMID 10720320. Bibcode 2000Sci...287.1995C.
- ↑ J. D. Lawrence; M. Takahashi; C. Bae; J. F. Hartwig. Regiospecific Functionalization of Methyl C−H Bonds of Alkyl Groups in Reagents with Heteroatom Functionality. Journal of the American Chemical Society. 2004, s. 15 334 – 15 335. DOI 10.1021/ja044933x. PMID 15563132.
- ↑ a b c J. F. Hartwig. Regioselectivity of the borylation of alkanes and arenes. Chemical Society Reviews. 2011, s. 1992–2002. DOI 10.1039/C0CS00156B. PMID 21336364.
- ↑ WEI, C. S.; JIMENEZ-HOYOS, C. A.; VIDEA, M.F.; HARTWIG, J. F.; HALL, M. B. Origins of the Selectivity for Borylation of Primary over Secondary C−H Bonds Catalyzed by Cp*-Rhodium Complexes. J. Am. Chem. Soc.. 2010, s. 3078–91. DOI 10.1021/ja909453g. PMID 20121104.
- ↑ Y. Kondo; D. Garcia-Cuadrado; J. F. Hartwig; N. K. Boaen; N. L. Wagner; M. A. Hillmyer. Rhodium-Catalyzed, Regiospecific Functionalization of Polyolefins in the Melt. Journal of the American Chemical Society. 2002, s. 1164–1165. DOI 10.1021/ja016763j. PMID 11841273.
- ↑ Carl N. Iverson; Milton R. Smith. Stoichiometric and Catalytic B−C Bond Formation from Unactivated Hydrocarbons and Boranes. Journal of the American Chemical Society. 1999-08-06, s. 7696–7697. DOI 10.1021/ja991258w.
- ↑ a b J. F. Hartwig. Borylation and silylation of C-H bonds: a platform for diverse C-H bond functionalizations. Accounts of Chemical Research. 2012, s. 864–873. DOI 10.1021/ar200206a. PMID 22075137.
- ↑ T. Ishiyama; J. Takagi; K. Ishida; N. Miyaura; N. Anastasi; J. F. Hartwig. Mild Iridium-Catalyzed Borylation of Arenes. High Turnover Numbers, Room Temperature Reactions, and Isolation of a Potential Intermediate. Journal of the American Chemical Society. 2002, s. 390–391. DOI 10.1021/ja0173019. PMID 11792205.
- ↑ Liskey, C. Iridium-Catalyzed Borylation of Aromatic and Aliphatic C–H bonds: Methodology and Mechanism. Dissertation, University of Illinois. Urbanan-Champaign. 2013
- ↑ FISCHER, D. F; SARPONG, R. Total Synthesis of (+)-Complanadine A Using an Iridium-Catalyzed Pyridine C−H Functionalization. Journal of the American Chemical Society. 2010, s. 5926–5927. DOI 10.1021/ja101893b. PMID 20387895.
- ↑ BOEBEL, T. A.; HARTWIG, J. F. Silyl-Directed, Iridium-Catalyzedortho-Borylation of Arenes. A One-Potortho-Borylation of Phenols, Arylamines, and Alkylarenes. Journal of the American Chemical Society. 2008, s. 7534–5. DOI 10.1021/ja8015878. PMID 18494474.
- ↑ ISHIYAMA, T.; MIYAURA, N.; ISOU, H.; KIKUCHI, T. Ortho-C–H borylation of benzoate esters with bis(pinacolato)diboron catalyzed by iridium–phosphine complexes. Chem. Commun.. 2010, s. 159–61. DOI 10.1039/b910298a. PMID 20024326.
- ↑ KAWAMORITA, S.; OHMIYA, H.; HARA, K.; FUKUOKA, A.; SAWAMURA, M. Directed Ortho Borylation of Functionalized Arenes Catalyzed by a Silica-Supported Compact Phosphine−Iridium System. J. Am. Chem. Soc.. 2009, s. 5058–9. DOI 10.1021/ja9008419. PMID 19351202.
- ↑ Ros, A.; Estepa, B.; Lopez-Rodriquez, R.; Alvarez, E.; Fernandez, R.; Lassaletta, J.M. Angew. Chem. Int. Ed. 2011; 50, 1.
- ↑ Boller, T.M.; Murphy, J. M.; Hapke, M.; Ishiyama, T.; Miyaura, N.; Hartwig, J.F. J. Am. Chem. Soc. 2005;, 127, 14263.
- ↑ T. A. Boebel; J. F. Hartwig. Silyl-Directed, Iridium-Catalyzedortho-Borylation of Arenes. A One-Potortho-Borylation of Phenols, Arylamines, and Alkylarenes. Journal of the American Chemical Society. 2008, s. 7534–7535. DOI 10.1021/ja8015878. PMID 18494474.
- ↑ R. Bisht; B. Chattopadhyay. Formal Ir-Catalyzed Ligand-Enabled Ortho and Meta Borylation of Aromatic Aldehydes via in Situ-Generated Imines. Journal of the American Chemical Society. 2016, s. 84–87. DOI 10.1021/jacs.5b11683. PMID 26692251.
- ↑ KANAI. A meta-selective C–H borylation directed by a secondary interaction between ligand and substrate. Nat. Chem.. 2015, s. 712–717. DOI 10.1038/nchem.2322. PMID 2 2629194 2. Bibcode 2015NatCh...7..712K.
- ↑ Akira Hirao; Shinichi Itsuno; Seiichi Nakahama; Noboru Yamazaki. Asymmetric reduction of aromatic ketones with chiral alkoxy-amineborane complexes. Journal of the Chemical Society, Chemical Communications. 1981, s. 315. DOI 10.1039/c39810000315.
- ↑ E. J. Corey; Raman K. Bakshi; Saizo Shibata. Highly enantioselective borane reduction of ketones catalyzed by chiral oxazaborolidines. Mechanism and synthetic implications. Journal of the American Chemical Society. 1987, s. 5551–5553. DOI 10.1021/ja00252a056.
- ↑ E. J. Corey; Raman K. Bakshi; Saizo Shibata; Chung Pin Chen; Vinod K. Singh. A stable and easily prepared catalyst for the enantioselective reduction of ketones. Applications to multistep syntheses. Journal of the American Chemical Society. 1987, s. 7925–7926. DOI 10.1021/ja00259a075.
- ↑ M. Mark Midland; Alfonso Tramontano; Stephen A. Zderic. The facile reaction of B-alkyl-9-borabicyclo[3.3.1]nonanes with benzaldehyde. Journal of Organometallic Chemistry. 1977, s. C17–C19. DOI 10.1016/S0022-328X(00)93625-8.
- ↑ M. Mark Midland; Alfonso Tramontano; Stephen A. Zderic. Preparation of optically active benzyl-.alpha.-d alcohol via reduction by B-3.alpha.-pinanyl-9-borabicyclo[3.3.1]nonane. A new highly effective chiral reducing agent. Journal of the American Chemical Society. 1977, s. 5211–5213. DOI 10.1021/ja00457a068.
- ↑ D. Ramesh; V. Shekhar; D. Chantibabu; S. Rajaram; U. Ramulu; Y. Venkateswarlu. First stereoselective total synthesis of pectinolide H. Tetrahedron Letters. 2012, s. 1258–1260. DOI 10.1016/j.tetlet.2011.12.122.
- ↑ Nicos A. Petasis; Irini Akritopoulou. The boronic acid mannich reaction: A new method for the synthesis of geometrically pure allylamines. Tetrahedron Letters. 1993, s. 583–586. DOI 10.1016/S0040-4039(00)61625-8.
- ↑ Tao Yu; Hui Li; Xinyan Wu; Jun Yang. Progress in Petasis Reaction. Chinese Journal of Organic Chemistry. 2012, s. 1836. DOI 10.6023/cjoc1202092.
- ↑ Thomas Herold; Reinhard W. Hoffmann. Enantioselective Synthesis of Homoallyl Alcohols via Chiral Allylboronic Esters. Angewandte Chemie International Edition in English. 1978, s. 768–769. DOI 10.1002/anie.197807682.
- ↑ William R. Roush; Alan E. Walts; Lee K. Hoong. Diastereo- and enantioselective aldehyde addition reactions of 2-allyl-1,3,2-dioxaborolane-4,5-dicarboxylic esters, a useful class of tartrate ester modified allylboronates. Journal of the American Chemical Society. 1985, s. 8186–8190. DOI 10.1021/ja00312a062.
- ↑ William R. Roush; Kaori Ando; Daniel B. Powers; Ronald L. Halterman; Alan D. Palkowitz. Enantioselective synthesis using diisopropyl tartrate modified (E)- and (Z)-crotylboronates: Reactions with achiral aldehydes. Tetrahedron Letters. 1988, s. 5579–5582. DOI 10.1016/S0040-4039(00)80816-3.
- ↑ William R. Roush; Paul T. Grover. Diisopropyl tartrate (E)-γ-(dimethylphenylsilyl)allylboronate, a chiral allylic alcohol β-carbanion equivalent for the enantioselective synthesis of 2-butene-1,4-diols from aldehydes. Tetrahedron Letters. 1990, s. 7567–7570. DOI 10.1016/S0040-4039(00)97300-3.
- ↑ William R. Roush; Paul T. Grover; Xiaofa Lin. Diisopropyl tartrate modified (E)-γ-[(cyclohexyloxy)dimethylsilyl-allylboronate, a chiral reagent for the stereoselective synthesis of anti 1,2-diols via the formal α-hydroxyallylation of aldehydes. Tetrahedron Letters. 1990, s. 7563–7566. DOI 10.1016/S0040-4039(00)97299-X.
- ↑ Rodney A. Fernandes; Pullaiah Kattanguru. Total synthesis of (8S,11R,12R)- and (8R,11R,12R)-topsentolide B2 diastereomers and assignment of the absolute configuration. Tetrahedron: Asymmetry. 2011, s. 1930–1935. DOI 10.1016/j.tetasy.2011.10.020.
- ↑ MIYAURA, Norio; SUZUKI, AKIRA. Stereoselective synthesis of arylated (E)-alkenes by the reaction of alk-1-enylboranes with aryl halides in the presence of palladium catalyst. Journal of the Chemical Society, Chemical Communications. 1979, s. 866. DOI 10.1039/C39790000866.
- ↑ MIYAURA, Norio; YAMADA, KINJI; SUZUKI, AKIRA. A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Letters. January 1979, s. 3437–3440. Dostupné online. DOI 10.1016/S0040-4039(01)95429-2.
- ↑ Gregor Nemecek; Robert Thomas; Helmut Goesmann; Claus Feldmann; Joachim Podlech. Structure Elucidation and Total Synthesis of Altenuic Acid III and Studies towards the Total Synthesis of Altenuic Acid II. European Journal of Organic Chemistry. 2013, s. 6420–6432. DOI 10.1002/ejoc.201300879.
- ↑ László Kürti; Barbara Czakó. Strategic applications of named reactions in organic synthesis : background and detailed mechanisms ; 250 named reactions. Amsterdam: Elsevier Academic Press, 2007. Dostupné online. ISBN 978-0-12-429785-2. S. 464–465.
- ↑ Tan D. Quach; Robert A. Batey. Copper(II)-Catalyzed Ether Synthesis from Aliphatic Alcohols and Potassium Organotrifluoroborate Salts. Organic Letters. 2003, s. 1381–1384. DOI 10.1021/ol034454n. PMID 12688764.
- ↑ Tan D. Quach; Robert A. Batey. Ligand- and Base-Free Copper(II)-Catalyzed C−N Bond Formation: Cross-Coupling Reactions of Organoboron Compounds with Aliphatic Amines and Anilines. Organic Letters. 2003-11-01, s. 4397–4400. DOI 10.1021/ol035681s. PMID 14602009.