
Aza-Copeův přesmyk
Aza-Copeův přesmyk je organická reakce, která má velké využití v organické syntéze, protože ji lze využít k rychlé tvorbě složitějších struktur ze strukturně jednodušších výchozích látek. Aza-Copeovy přesmyky patří mezi heteroatomární varianty Copeova přesmyku,což je [3,3]-sigmatropní přesmyk, při němž se posouvají jednoduché a dvojné vazby mezi dvěma allylovými reaktanty. Podle Woodwardových–Hoffmannových pravidel probíhají tepelné aza-Copeovy přesmyky suprafaciálně.[1] Aza-Copeovy přesmyky se klasifikují podle polohy dusíku v molekule:
![[1,2] a [1,3] aza-Copeovy přesmyky](http://upload.wikimedia.org/wikipedia/commons/thumb/e/ee/Aza-cope_intropicture_final.tiff/lossy-page1-568px-Aza-cope_intropicture_final.tiff.jpg)
Prvním popsaným případem aza-Copeova přesmyku byl kationtový 2-aza-Copeův přesmyk, probíhající za teploty o 100 až 200 °C nižší než Copeův přesmyk.[2] Na snadném provedení reakce má podíl termoneutralita reakce a přítomnost kladně nabitého dusíku v molekule, která snižuje její aktivační energii. K méně častým reakcím patří 1-aza- a 3-aza-Copeovy přesmyky, které mají vyšší aktivační energie a omezenější syntetické využití.[3][4][5] Za účelem vylepšení využitelnosti se kationtové 2-aza-Copeovy přesmyky obvykle spojují s dalšími reakcemi, jako je Mannichova cyklizace. Tato spojená reakce probíhá za mírných podmínek, vyzbačuje se diastereoselektivitou, a má velké syntetické využití. Lze pomocí ní snadno připravit acylované pyrrolidiny, což je struktura nacházející se v řadě přírodních látek, jako jsou například alkaloidy, a používá se například při přípravě strychninu a krininu.[6] Aza-Copeovými přesmyky se podrobně zabýval Larry E. Overman.[1]
Kationtový 2-aza-Copeův přesmyk[editovat | editovat zdroj]

Kationtový 2-aza-Copeův přesmyk, přesněji nazývaný jako 2-azonia-[3,3]-sigmatropní přesmyk, studoval Larry E. Overman. Jedná se o nejlépe prozkoumaný druh aza-Copeova přesmyku, protože probíhá za mírných podmínek a má také široké syntetické využití, například při syntéze alkaloidů. Při 2-aza-Copeových přesmycích nevznikají vedlejší produkty, jelikož štěpení a tvorba vazeb v obou směrech jsou ekvivalentní, podobně jako u Copeova přesmyku. Přítomnost nabitého atomu dusíku způsobuje, že je reakce oproti klasickému Copeovu přesmyku rychlejší.
První 2-aza-Copeův přesmyk byl popsán v roce 1950, a to při neúspěšném pokusu o přípravu aminoalkoholu.[2] This discovery identified the basicByl určen základní mechanismus přesmyku podle toho, že produkt se pravděpodobně vytvořil při dusíkatém ekvivalentu Copeova přesmyku. Reakcí allylbenzylaminu (A) s kyselinou mravenčí a formaldehydem vznikl aminoalkohol (B). Aminoalkohol se adicí kyseliny přeměnil na imin (C), u kterého proběhl kationtový 2-aza-Copeův přesmyk (D). Hydrolýzou iminiového iontu vznikl amin (E). Při reakci výchozí látky se samotným formaldehydem po přesmyku docházelo k alkylaci aminové skupiny.[2]

Jelikož reakce, na rozdíl od čistě uhlovodíkového Copeova přesmyku, probíhá za mírných podmínek, tak se předpokládá, že kladný náboj na dusíku výrazně snižuje aktivační energii přesmyku.[2]
Mechanismus[editovat | editovat zdroj]
Urychlení reakce kladně nabitým dusíkem[editovat | editovat zdroj]
Aza-Copeovy přesmyky podle Woodwardových–Hoffmannových pravidel probíhají suprafaciálně. Toto nebylo ověřeno a takový průběh se pouze předpokládá na základě skutečnosti, že při zásaditě katalyzovaném oxy-Copeově přesmyku nabitý atom narušuje sigmatropní přesmyk a mění mechanismus ze soustředěného (který by se u Copeova přesmyku dal očekávat) na částečně diradikálový/dipolární, což je způsobeno delokalizací kladného náboje na allylovém zbytku, což vede k oslabení allylové vazby a snížení aktivační energie štěpení vazby. Kationtové aza-Copeovy přesmyky tak probíhají rychleji než klasické Copeovy přesmyky.[6][7]
Meziprodukt a stereochemie[editovat | editovat zdroj]
Kationtové 2-aza-Copeovy přesmyky se vyznačují vysokou stereospecificitou, která je důsledkem toho, že u meziproduktu výrazně převažuje židličková konformace nad ostatními. L. E. Overman se svými spolupracovníky podrobně prozkoumali tuto vlastnost podobnými postupy, jako u Doeringova a Rothova experimentu,[8] který ukázal na převahu židličkové konformace.[9] S využitím kationtového 2-aza-Copeovy/Mannichovy reakce pyrrolizidinů zjistili, že většinově vznikají produkty s cis substituenty u E-alkenových a s trans substituenty u Z-alkenových reaktantů, což naznačuje tvorbu meziproduktu v židličkové konformaci. Kdyby vznikal přechodný stav s lodičkovou konformací, tak by reakce probíhala opačně.[9] Podobně jako u mnoha jiných reakcí má přeměna Z-enolátů kvůli 1,3-diaxiálním sterickým interakcím mezi enolátem a cyklem nižší selektivitu. Tímto lze vysvětlit potřebu vyšších teplot při reakcích Z-enolátů.[6][9] U kationtových 2-aza-Copeových přesmyků je přechodný stav v lodičkové konformaci ještě méně pravděpodobný než u klasických Copeových přesmyků: podobně v případech, kdy Copeovy přesmyky mají lodičkové přechodné stavy, tak meziprodukty aza-Copeových přesmyků stále mají židličkovou geometrii.[1][6][10] Tyto výsledky odpovídají údajům získaným pomocí výpočetních metod.[11]

Výsledky těchto experimentů také naznačují, že kationtové 2-aza Copeovy přesmyky (stejně jako Mannichovy cyklizace) probíhají rychleji než enolové či iminiové tautomerizace. Pokud by to mu tak nebylo, tak by se neobjevovala žádná stereospecifita.[1]
Další faktory ovlivňující stereochemii[editovat | editovat zdroj]
Na stereochemii aza-Copeových/Mannichových reakcí použitých v anulačních reakcích má vliv meziprodukt s židličkovou konformací, který způsobuje, že objemné substituenty se obvykle navážou kvaziekvatoriálně. Vinylové a aminové skupiny mohou reakci řídit podle toho, zda jsou v konfiguraci syn- nebo anti. Velký vliv mívají aminové skupiny, u kterých velké substituenty vedou k tvorbě syn-produktů. U reaktantů s anti vinylovými a aminovými substituenty vzniká jediný přechodný stav, vedoucí ke vzniku cis cyklických sloučenin, zatímco v případě syn substituentů mohou vznikat různé meziprodukty v závislosti na sterických interakcích s rozpouštědlem nebo velkými dusíkatými substituenty, u nichž může dojít ke změnám konfigurace přechodného stavu.[12][13]
U jednoduchých aza-Copeových/Mannichových reakcí nezařazených do anulací, jako jsou kondenzace aminoalkoholů s ethery, probíhají rotace vazeb rychleji než Mannichovy cyklizace a tvoří se racemáty.[14] Tomu se lze vyhnout použitím chirálních pomocníků v podobě substituentů na aminových skupinách. U takto upravených reaktantů k rotacím vazeb nedochází.[1]

Aza-Copeova/Mannichova reakce[editovat | editovat zdroj]
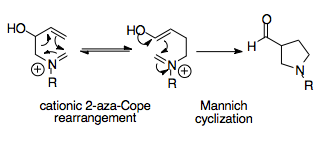
Aza-Copeova/Mannichova reakce je synteticky dobře využitelná reakce, pomocí které lze získat složité cyklické molekuly z jednoduchých výchozích látek. Umožňuje termodynamické řízení ve prospěch konkrétního produktu, protože Mannichova cyklizace je nevratná a její produkt, acylovaný pyrrolidin, vykazuje větší stabilitu než produkt přesmyku.[1][15]
První aza-Copeova/Mannichova reakce[editovat | editovat zdroj]
L. E. Overman se svými spolupracovníky zjistil, že kationtové 2-aza-Copeovy přesmyky by mohly být synteticky využitelné, pokud by bylo možné využít vhodný způsob termodynamického řízení. Tato myšlenka byla využita k zapojení nukleofilních substituentů, konkrétně alkoholových skupin, do výchozích látek, které se po provedení přesmyku přemění na enoly, čímž umožní atak iminiového iontu.
První popsanou variantou byla reakce aldehydehydů s 2-alkoxy-3-butenaminy, kterou vznikaly aminoalkoholy, jejichž aza-Copeovy/Mannichovy produkty byly acylované pyrrolidiny. Tento postup vyžadoval mírné zahřívání po dobu několika hodin. Ukázalo se, že aza-Copeovy/Mannichovy reakce se vyznačují velmi dobrými výtěžnostmi. Lze je lehce využít ke kondenzačním reakcím aminoetherů, při kterých se nejprve methyluje alkohol.[15] Po aza-Copeově/Mannichově reakci se adicí za přítomnosti hydroxidu sodného vytvoří keton.[15] Amin zde nemůže způsobit tvorbu iminiových iontů ze zásaditých ketonů; metody zapojující do reakce ketony byly vyvinuty později.[15][16] Výhodnost této reakce mimo jiné dokazuje to, že reakce probíhá, i když se vytvoří méně stabilní izomer, z čehož vyplývá termodynamická výhodnost reakce.[12][16]

Mechanismus[editovat | editovat zdroj]
Hlavní produkt reakce může vznikat dvěma různými způsoby: aza-Copeovou/Mannichovou reakcí a aza-Prinsovou cyklizací s pinakolovým přesmykem. Tyto mechanismy mají rozdílné stereochemické vlastnosti, čímž lze vysvětlit převahu aza-Copeova/Mannichova mechanismu. Při aza-Copeově/Mannichově reakci každý atom v analogu [1,5] dienu podléhá sp2 hybridizaci, čímž naruší stereochemii reaktantu na pozici R', zatímco u aza-Prinsovy reakce s pinakolovým přesmykem zůstává stereochemie na tomto místě zachována; díky tomu je možné pomocí jednoduchého testu zjistit, který mechanismus převažuje. U enantiomerně čistého výchozího materiálu v pozici "R'" bude vznikat racemická směs, pokud je převažujícím mechanismem aza-Copeova/Mannichova reakce, zatímco při převaze aza-Prinsovy cyklizace s pinakolovým přesmykem by stereochemie měla být zachována. Zda je produkt racemický, lze ověřit jednoduše, díky čemuž se dá ověřit, že převažujícím mechanismem je aza-Copeova/Mannichova reakce. Tato skutečnost byla ověřena tím, že účinnost tvorby karbeniového iontu při aza-Prinsově/pinakolovém mechanismu by bylo možné zvýšit stabilizací kladného náboje vhodným substituentem, který by pozměnil reaktivitu při tomto průběhu. Bylo vyzkoušeno několik různých substituentů a zjistilo se, že jejich vliv na výsledek reakce je malý, což také ukazuje na převahu aza-Copeova/Mannichova mechanismu.[14] Aza-Prinsův/pinakolový mechanismus se projevuje pouze při výrazně navýšené nukleofilitě alkenu a elektrofilitě iminu.[1][6][17]
Aza-Copeova/Mannichova reakce má vysokou diastereoselektivitu, což je v souladu s výsledky stereochemických experimentů, při kterých byly určeny vlastnosti meziproduktu kationitového 2-aza-Copeova přesmyku. Stereochemie této reakce je poněkud složitější, pokud jsou na cykly v reaktantech navázány allylové a aminové substituenty, kdy se projevuje i cis-trans izomerie.
Využití 2-aza-Copeovy/Mannichovy reakce[editovat | editovat zdroj]
Aza-Copeovy/Mannichovy reakce jsou často nejefektivnějšími způsoby syntézy pyrrolidinů a využívají se tak při přípravě mnoha přírodních látek. Díky její diastereoselektivitě lze tuto reakci použít k asymetrické syntéze, například při přípravě alkaloidů. Může být použita k tvorbě dalších cyklických struktur využitelných v syntézách, například indolizidinů a indolů.[1]
Totální syntéza (−)-strychninu[editovat | editovat zdroj]
Praktické využití 2-aza-Copeovy/Mannichovy reakce lze ukázat na příkladu Overmanovy totální syntézy strychninu, což je alkaloid nacházející se v rostlinách z rodu kulčiba (Strychnos). Strychnin je často používanou látkou hubící malé obratlovce. První totální syntézu strychninu popsal R. B. Woodward,[18] přičemž do té doby se nepodařilo uměle vytvořit sloučeninu s takto složitou strukturou. Další totální syntézy byly popsány koncem 80. let 20. století; při nich byly používány podobné postupy, jako jsou využití meziproduktů, které také vznikají rozkladem strychninu. Všechny tyto syntézy probíhaly za tvrdých podmínek. Overman tyto potíže překonal a vyvinul i první asymetrickou totální syntézu strychninu, čímž dosáhl diastereoselektivity a mírných reakčních podmínek, při jakých se provádí aza-Copeova/Mannichova reakce. Krok zahrnující aza-Copeovu/Mannichovu reakci měl téměř stoprocentní výtěžnost. Overmanova syntéza je o několik řádů účinnější než předchozí postupy.[18]

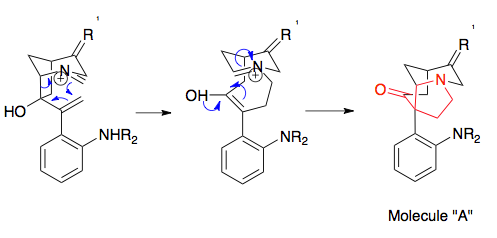
Overmanovu syntézu strychninu lze považovat za dobrý příklad přípravy prekurzorů potřebných pro aza-Copeovu/Mannichovu reakci, protože se jedná o účinné využití otevírání epoxidového kruhu. Hlavní kroky syntézy substrátu zahrnují Stilleovu reakci, při níž se spojí dva prekurzory, epoxidaci dvojné vazby terc-butylhydroperoxidem, Wittigovu reakci sloužící k přeměně ketonu na alken, a cyklizaci. Následně se provede alkylace aminu (není znázorněna), čímž se vytváří substrát pro přesmyk. Tento postup se vyznačuje enantioselektivitou podobnou jako u aza-Copeovy/Mannichovy reakce, protože je konfigurace konečného produktu určena konfigurací výchozí látky: který enantiomer strychninu se vytvoří, je určeno enantiomerem výchozího materiálu.[18][19]

Syntéza (−)-krininu[editovat | editovat zdroj]
Krinin je alkaloid vyskytující se u rostlin z čeledi amarylkovitých a jedna z prvních látek připravená pomocí asymetrické totální syntézy s využitím aza-Copeovy/Mannichovy reakce. Jednalo se o významný pokrok ve výzkumu aza-Copeových/Mannichových reakcí, jelikož patří mezi jejich nejvyužívanější varianty. Využívá vlastnosti kationtových-2-aza-Copeových přesmyků v podobě vysoké diastereoselektivity a také se při ní používají kyanomethylové chránicí skupiny, které chrání aminové skupiny v průběhu adice vinyllithia; jako odstupující skupina rovněž vyvolávají vznik iminiových iontů, asistovaný adicí dusičnanu stříbrného.[20]
Tato syntéza je jednou z mnoha syntetických metod, které využívají kyanomethylové skupiny k přípravě pyrrolidinů a indolizidinů.

Příprava tricyklických alkaloidů[editovat | editovat zdroj]
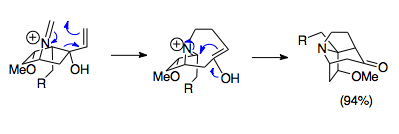
Overman se svými spolupracovníky vyvinul řadu postupů syntézy složitých tricyklicých struktur pomocí aza-Copeových/Mannichových reakcí. Tyto aza-tricyklické struktury jsou součástmi alkaloidů vyskytujících se u rostlin rodu Stemona a mohou mít uplatnění při výrobě některých léčiv, například imunosupresiv. Na následujícím obrázku je jako příklad znázorněna reakce 1-aza-bicyklo[2.2.1]heptanové soli s paraformaldehydem při 80 °C vedoucí ke vzniku pivotalové aza-tricyklické skupiny. I přes nevýhodnost překryvu orbitalů v důsledku sterických efektů se dosahuje výtěžnosti o hodnotě 94 %.[21]
Otevírání a rozšiřování cyklů[editovat | editovat zdroj]

Aza-Copeovy/Mannichovy reakce, kterých se účastní molekuly již obsahující cykly, lze použít k přípravě indolizidinových (s pyrolidinem spojeným s cyklohexanovým kruhem) sloučenin. Při těchto reakcích se otevírají cyklopentanové kruhy pomocí přesmyků a uzavírají se Mannichovou cyklizací za vzniku šestičlenných cyklů navázaných na pyrrolidinovou skupinu. Takto je možné získat i sedmičlenné cykly, protože enoly a iminiové ionty se nacházejí v dostatečné blízkosti, aby u nich mohla proběhnout Mannichova cyklizaci.[20] Příprava makrocyklů tímto způsobem nebyla popsána, protože enol a iminium se nenacházejí dostatečně blízko u sebe.[6]
Jako substráty přesmyku lze použít i vinyloxazolidiny. Nejprve se tvoří vinyloxazolidin atakem aminobutenolu na cyklohexanon a následně dojde při zahřívání za přítomnosti kyseliny (Lewisovy nebo protické) aza-Copeova/Mannichova reakce. Poté proběhne rozpad cyklu a tvorba pětičlenného kruhu. Další možností je navázání oxazolidinu na další kruh.[22]
Srovnání s jinými metodami[editovat | editovat zdroj]
Aza-Copeovy/Mannichovy reakce mají řadu výhod ve srovnání s jinými metodami; patří k nim nízká reakční teplota (obvykle do 80 °C), velké množství použitelných rozpouštědel a přídavek 1 ekvivalentu kyseliny, kterou je obvykle kyselina kamforsulfonová nebo některá Lewisova kyselina. Ostatní způsoby přípravy pyrrolidinů nemají takovou stereospecificitu, široké využití, ani spektrum použitelných výchozích látek. Reakce má vysokou diastereoselektivitu a probíhá i v případech, kdy je v přechodném stavu nevýhodný překryv orbitalů.[1]

Tyto vlastnosti aza-Copeovy/Mannichovy reakce vedly k hledání způsobů přípravy vhodných výchozích látek; tyto metody se dělí do dvou hlavních oblastí: adice aminů s tvorbou iminiových iontů (znázorněna červeně) a navazování vinylových substituentů (znázorněno modře). Při reakci může být použito mnoho různých N-substituentů (R), alkylových i arylových, přičemž některé ovlivňují stereochemii produktů. Z vinylových skupin jsou obvykle vhodné jen ty, které jsou 1,1- nebo 1,2-disubstituované.[1]
Adice aminů s tvorbou iminiových iontů[editovat | editovat zdroj]
Otevírání epoxidových kruhů[editovat | editovat zdroj]
Díky úhlovému napětí lze na molekuly epoxidů snadno navázat aminové skupiny dva atomy od alkoholových skupin. Epoxidový kruh je nejdříve otevřen nukleofilním atakem bromidu; jako nukleofily mohou rovněž sloužit primární nebo aromatické aminy či lithné anilidy. Po tomto kroku se často provádí O-methylace, čímž se naváže chránicí skupina.

Pokud sterické efekty umožní atak pouze na jednom konkrétním uhlíku, tak nastane přímý vnitromolekulární atak na dusík; taková reakce probíhá například při syntéze strychninu.[15][23]
Tvorba iminiových iontů[editovat | editovat zdroj]
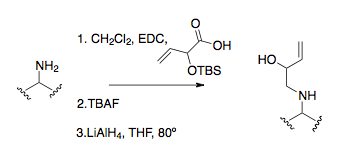
Nejčastějším způsobem tvorby iminiových iontů při napojování aminových skupin je adice formaldehydu nebo paraformaldehydu, kde se iminium tvoří kysele katalyzovanou kondenzací. Tento postup je mimo jiné součástí Overmanovy syntézy strychninu.[6][23] Občas se využívají též vnitromolekulární reakce s karbonylovými skupinami.[9] Dalšími možnostmi jsou využití kyanomethylových skupin a nebo oxazolidinových prekurzorů karbonylů.
Alkylace aminů[editovat | editovat zdroj]
Alkylace aminů je častou metodou přípravy prekurzorů iminů. Alkylace přímými SN2 reakcemi se provádí jen ojediněle, protože u aminů často dochází k přealkylování, tedy navázání vyššího počtu alkylů, než jaký je potřeba.[23] Častější je reduktivní aminace, která byla použita i u prvního aza-Copeova přesmyku.[15][24][25]
Nejčastějším způsobem alkylace aminů je tvorba amidové vazby a její následná redukce, obvykle pomocí hydridu lithno-hlinitého.[9]
Použití oxazolidinů[editovat | editovat zdroj]
Ketony a stericky ovlivněné aldehydy nelze zapojit do zásaditě katalyzovaných aza-Copeových/Mannichových reakcí, protože aminy s nimi nemohou vytvářet iminiové ionty. Náhradou je dehydratační tvorba oxazolidinů nanásledovaná zahříváním za přítomnosti stechiometrického množství kyseliny.
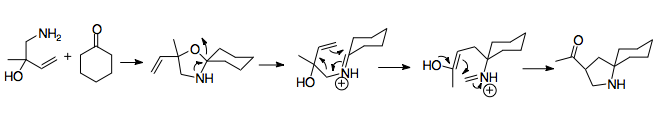
Využití oxizolidinů k získání iminiových iontů popsal L. E. Overman; zjistil, že při přípravě pyrrolidinů lze jako karbonylové reaktanty použít deriváty cyklohexanonu.[16] Je možné použít mnoho různých cyklohexanonů. Při použití substituovaných acyklických ketonů má reakce kvůli termodynamické výhodnosti odštěpení cyklohexanonu, způsobené úhlovým napětím v židličkové konformaci, nízkou výtěžnost.
Pomocí oxazolidinů se například vytváří 1-azaspiro[4,5]dekanové cykly, používané při přípravách přírodních látek.[16]
[editovat | editovat zdroj]
Vinylace ketonů[editovat | editovat zdroj]
Vinylací lze získat další syntetické možnosti, a tím rozšířit využitelnost reakce.[21] Obvykle se k tomuto účelu používají organolithné sloučeniny. Často se také na dusík připojuje substituent nebo chránicí skupina, i když to není vždy nutné. Přidání lithia do reakce má výrazný vliv na stereochemii výchozího materiálu, protože se na něj koordinuje dusík. Z takto upravených výchozích materiálů obvykle vznikají anti aza-Copeovy prekurzory, zatímco z ostatních, například těch, které obsahují vysoce substituované a stericky ovlivněné aminy, se tvoří syn prekurzory. Tyto vlivy dusíkatých substituentů mají velký význam.[6][23]
Využití kyanomethylových skupin[editovat | editovat zdroj]
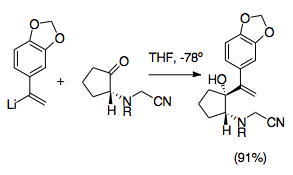
Kyanomethylové skupiny jsou snadným způsobem ochrany iminiových iontů během allylové vinylace ketonů. Kyanamidové skupiny a jejich analogy se při přípravě iminiových iontů využívají často. Obvykle se navazují prostřednictvím nukleofilních adicí na iminium, nejčastěji alkylací aminu formaldehydem. Iminiový ion je tak stíněn.[26] Použití kyanomethylových skupin umožňuje snadné řízení aza-Copeovy/Mannichovy reakce. Kyanomethyl chrání dusík v pozici 2 v průběhu tvorby dalších allylových analogů. Následně představuje dobrou odstupující skupinu při vzniku iminiového iontu.[27]
Vznik iminiových iontů z kyanomethylových skupin se obvykle spouští přídavkem dusičnanu stříbrného, lze však také použít i jiné sloučeniny stříbra a nebo mědi. Tímto způsobem lze řídit tvorbu iminiových iontů s ještě větší přesností.[6][27] Uvedené reakce se musí provádět za teplot okolo −78 °C, aby nedocházelo ke vzájemnému působení mezi kyanomethyly a vinyllithiem. Tato metoda umožňuje využití mnoha různých dusíkatých substituentů a může být použita ke zjednodušení syntézy oktahydroindolů a pyrrolů.[1][27]

1- a 3-aza-Copeovy přesmyky[editovat | editovat zdroj]

1- a 3-aza-Copeovy přesmyky jsou ve srovnání s kationtovými 2-aza-Copeovými přesmyky vzácnější, jelikož mají mnohem vyšší aktivační energie.
1- a 3-aza-Copeovy přesmyky vedou ke vzniku iminů častěji než k enaminům, protože vazby π mezi uhlíkem a dusíkem jsou silnější než příslušné vazby σ, takže je termodynamicky výhodnější 3-aza-přesmyk oproti 1-aza-přesmyku: imin má energii o asi 40 kJ/mol nižší. Vysoké aktivační energie těchto reakcí jsou tak kinetického původu.[28] Výzkumy 1- i 3-aza-Copeových přesmyků byly zaměřené na hledání způsobů snížení těchto aktivačních energií. Pro možné syntetické využití bylo vyvinuto několik variant takovýchto přesmyků. 1-aza-Copeovy přesmyky se obvykle propojují s termodynamickými metodami a 3-aza-Copeovy přesmyky se většinou provádějí kationtově.[28][29]
Jak 1-, tak i 3-aza-Copeovy přesmyky probíhají převážně přes meziprodukty s židličkovými konformacemi (a zachovává se při nich stereochemie, podobně jako u kationtových 2-aza-Copeových přesmyků), lze je urychlit pomocí zavedení kladných nábojů, což dodává meziproduktům více diradikálové a dipolární vlastnosti.[29] 3-aza-Copeovy přesmyky (a tedy i 1-aza-přesmyky, které probíhají přes stejné přechodné stavy) by tak měly mít méně aromatické přechodné stavy než obecné Copeovy přesmyky a kationtové 2-aza-Copeovy přesmyky, což vede k vyšším reakčním teplotám (blízkým teplotám využívaným u Copeových přesmyků i vyšších, mezi 170 a 300 °C) potřebným k překonání kinetických aktivačních bariér.[3][29][30]
3-aza-Copeovy přesmyky[editovat | editovat zdroj]

3-aza-Copeův přesmyk byl objeven krátce po 2-aza-Copeově přesmyku, protože má podobný vztah ke Claisenově přwsmyku; nejprve byl popisován jako amino-Claisenův přesmyk, což je nepřesné označení, protože molekuly, které se do něj zapojují, obsahují atomy dusíku i kyslíku.[3] Tento přesmyk se používá na přípravu heterocyklů obsahujících uhlík, nejčastěji piperidinů.
Jeden z prvních případů popsal Robert Burpitt, který si všiml přesmyku probíhajícího u kvartérních amoniových solí, kde v důsledku působení elektrického náboje, probíhal exotermicky, aniž by bylo nutné zahřívání; bez přítomnosti tetrasubstituovaného dusíku by k reakci nedošlo.[31] Na základě tohoto objevu byl výzkum 3-aza-Copeovýcgh přesmyků zaměřen převážně na zwitteriontové varianty reakce, protože rozprostření elektrického náboje snižuje aktivační energii: v některých případech lze reakci provést i při teplotách okolo −20 °C.[28][32]
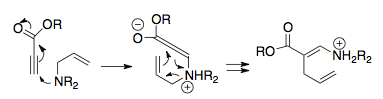
První 3-aza-Copeův přesmyk nenabitých molekul byl proveden v roce 1967. Po vytvoření dostatečně substituovaných enaminů a následném zahřívání proběhl téměř úplný přesmyk na iminy; tato reakce má však jen omezené možnosti využití.[33]
1-aza-Cope rearrangement[editovat | editovat zdroj]
První popsaný 1-aza-Copeův přesmyk byl analogem obecného Copeova přesmyku a k jeho provedení bylo třeba zahřívání na vysoké teploty, aby byla překonána vysoká termodynamická aktivační bariéra; většina dalšího výzkumu ohledně 1-aza-Copeových přesmyků tak byla soustředěna na zmírnění reakčních podmínek. V důsledku přítomnosti heteroatomu se v průběhu reakce v kroku určujícím její rychlost pravděpodobně vytváří meziprodukt s částečně diradikálovými a dipolárními vlastnostmi.[4]
F. W. Fowler učinil z 1-aza-Copeových přesmyků postupy prakticky využitelné v organické syntéze.[3] Všiml si, že vysoká energetická bariéra reakce způsobuje, že produkt přetrvává v iminové formě, a tak by mohlo být výhodné dusík stabilizovat. Za tímto účelem napojil na dusík karbonylovou skupinu, protože volný elektronový pár na dusíku by měl být stabilizován účastí na amidové vazbě, a tak být elektronegativita této skupiny nižší než u LUMO iminové skupiny, což by vedlo k upřednostnění přechodného stavu.[3] Tímto způsobem se podařilo pomocí 1-aza-Copeova přesmyku připravit deriváty piperidinu a pyridinu. Ukázalo se, že lze takto připravovat i ty produkty, u nichž reakce musí probíhat přes lodičkový meziprodukt nebo společně s jinými přesmyky.[3] Důležitá je též poměrně snadná příprava reaktantů, při kterých se používají Dielsovy–Alderovy reakce.[3]
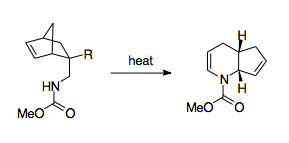
Dalším způsobem překonání termodynamické bariéry je propojení reakce s uvolněním napětí na cyklopropanovém kruhu, které dovoluje výrazně snížit reakční teplotu.[28][34]

Reference[editovat | editovat zdroj]
- ↑ a b c d e f g h i j k L. E. Overman; P. G. Humphreys; G. S. Welmaker. Organic Reactions. [s.l.]: [s.n.], 2011. ISBN 978-0471264187. DOI 10.1002/0471264180.or075.04. Kapitola The Aza-Cope/Mannich Reaction, s. 747–820.
- ↑ a b c d R. M. Horowitz; T. A. Geissman. A Cleavage Reaction of α-Allylbenzylamines. Journal of the American Chemical Society. 1950, s. 1518–1522. DOI 10.1021/ja01160a025.
- ↑ a b c d e f g M. Chu; P. L. Wu; S. Givre; F. W. Fowler. The 1-AZA-Cope rearrangement. Tetrahedron Letters. 1986, s. 461–464. DOI 10.1016/S0040-4039(00)85505-7.
- ↑ a b P. L. Wu; F. W. Fowler. The 1-aza-Cope rearrangement. 2. The Journal of Organic Chemistry. 1988, s. 5998–6005. DOI 10.1021/jo00261a003.
- ↑ G. R. Cook; N. S. Barta; J. R. Stille. Lewis acid-promoted 3-aza-Cope rearrangement of N-alkyl-N-allyl enamines. The Journal of Organic Chemistry. 1992, s. 461–467. DOI 10.1021/jo00028a016.
- ↑ a b c d e f g h i L. E. Overman; L. T. Mendelson; E. J. Jacobsen. Synthesis applications of aza-Cope rearrangements. 12. Applications of cationic aza-Cope rearrangements for alkaloid synthesis. Stereoselective preparation of cis-3a-aryloctahydroindoles and a new short route to Amaryllidaceae alkaloids. Journal of the American Chemical Society. 1983, s. 6629–6637. DOI 10.1021/ja00360a014.
- ↑ L. E. Overman. Charge as a key component in reaction design. The invention of cationic cyclization reactions of importance in synthesis. Accounts of Chemical Research. 1992, s. 352–359. DOI 10.1021/ar00020a005.
- ↑ W. v. E. Doering; W. R. Roth. The overlap of two allyl radicals or a four-centered transition state in the cope rearrangement. Tetrahedron. 1962, s. 67–74. DOI 10.1016/0040-4020(62)80025-8.
- ↑ a b c d e R. J. Doedens; G. P. Meier; L. E. Overman. Synthesis applications of cationic aza-Cope rearrangements. Part 17. Transition-state geometry of [3,3]-sigmatropic rearrangements of iminium ions. The Journal of Organic Chemistry. 1988, s. 685–690. DOI 10.1021/jo00238a039.
- ↑ E. Vogel; W. Grimme; E. Dinne. Thermal Equilibrium between cis-1,2-Divinylcyclo-pentane and cis,cis-1,5-Cyclononadiene. Angewandte Chemie International Edition in English. 1963, s. 739–740. DOI 10.1002/anie.196307392.
- ↑ M. Lukowski; K. Jacobs; P. Hsueh; H. A. Lindsay; M. C. Milletti. Thermodynamic and kinetic factors in the aza-Cope rearrangement of a series of iminium cations. Tetrahedron. 2009, s. 10311–10316. DOI 10.1016/j.tet.2009.10.010.
- ↑ a b S. F. McCann; L. E. Overman. Medium Effects and the Nature of the Rate-Determining Step in Mannich-Type Cyclizations. Journal of the American Chemical Society. 1987, s. 6107–6114. DOI 10.1021/ja00254a033.
- ↑ L. E. Overman; W. C. Trenkle. Controlling Stereoselection in Aza-Cope-Mannich Reactions. Isr. J. Chem.. 1997, s. 23–30. DOI 10.1002/ijch.199700005.
- ↑ a b E. J. Jacobsen; J. Levin; L. E. Overman. Synthesis applications of cationic aza-Cope rearrangements. Part 18. Scope and mechanism of tandem cationic aza-Cope rearrangement-Mannich cyclization reactions. Journal of the American Chemical Society. 1988, s. 4329–4336. DOI 10.1021/ja00221a037.
- ↑ a b c d e f L. E. Overman; M. Kakimoto. Carbon-Carbon Bond Formation via Directed 2-Azonia-[3,3]-Sigmatropic Rearrangements. A New Pyrrolidine Synthesis. Journal of the American Chemical Society. 1979, s. 1310–1312. DOI 10.1021/ja00499a058.
- ↑ a b c d L. E. Overman; M. Kakimoto; M. Kakimoto. Directed 2-azonia-[3,3]-sigmatropic rearrangements. a convenient preparation of substituted 1-azaspiro[4,5]decanes. Tetrahedron Letters. 1979, s. 4041–4044. DOI 10.1016/s0040-4039(01)86498-4.
- ↑ A. Armstrong; J. Levin; L. E. Overman. aza-Prins-pinacol approach to 7-azabicyclo[2.2.1]heptanes and ring expansion to [3.2.1]tropanes. Organic Letters. 2005, s. 1335. DOI 10.1021/ja00221a037.
- ↑ a b c d R. B. Woodward; M. P. Cava; W. D. Ollis; A. Hunger; H. U. Daeniker; K. Schenker. The total synthesis of strychnine. Tetrahedron. 1963, s. 247–288. DOI 10.1016/S0040-4020(01)98529-1. PMID 13305562.
- ↑ S. D. Knight; L. E. Overman; G. Pairaudeau. Synthesis applications of cationic aza-Cope rearrangements. 26. Enantioselective total synthesis of (−)-strychnine. Journal of the American Chemical Society. 1993, s. 9293–9294. DOI 10.1021/ja00073a057.
- ↑ a b L. E. Overman; S. Sugai. Total Synthesis of (−)-Crinine. Use of Tandem Cationic Aza-Cope Rearrangement/Mannich Cyclizations for the Synthesis of Enantiomerically Pure Amaryllidaceae Alkaloids. Helvetica Chimica Acta. 1985, s. 745–749. DOI 10.1002/hlca.19850680324.
- ↑ a b M. Brueggemann; A. I. McDonald; L. E. Overman; M. D. Rosen; L. Schwink; J. P. Scott. Total Synthesis of (±)-Didehydrostemofoline (Asparagamine A) and (±)-Isodidehydrostemofoline. Journal of the American Chemical Society. 2003, s. 15 284 – 15 285. DOI 10.1021/ja0388820. PMID 14664560.
- ↑ L. E. Overman; J. Shim. Synthesis applications of cationic aza-Cope rearrangements. Part 25. Total synthesis of Amaryllidaceae alkaloids of the 5,11-methanomorphanthridine type. Efficient total syntheses of (−)-pancracine and (.+-.)-pancracine. Organic Reactions. 1993, s. 4662–4672. DOI 10.1021/jo00069a032.
- ↑ a b c d L. E. Overman; M. Kakimoto; M. E. Okazaki; G. P. Meier. Synthesis applications of aza-Cope rearrangements. 11. Carbon-carbon bond formation under mild conditions via tandem cationic aza-Cope rearrangement-Mannich reactions. A convenient synthesis of polysubstituted pyrrolidines. Journal of the American Chemical Society. 1983, s. 6622–6629. DOI 10.1021/ja00360a013.
- ↑ L. E. Overman; C. Fukaya. Stereoselective total synthesis of (.+-.)-perhydrogephyrotoxin. Synthetic applications of directed 2-azonia-[3,3]-sigmatropic rearrangements. Journal of the American Chemical Society. 1980, s. 1454–1456. DOI 10.1021/ja00524a057.
- ↑ R. F. Borch; M. D. Bernstein; H. D. Durst. Cyanohydridoborate anion as a selective reducing agent. Journal of the American Chemical Society. 1971, s. 2897–2904. DOI 10.1021/ja00741a013.
- ↑ D. S. Grierson; M. Harris; H. P. Husson. Synthesis and chemistry of 5,6-dihydropyridinium salt adducts. Synthons for general electrophilic and nucleophilic substitution of the piperidine ring system. Journal of the American Chemical Society. 1980, s. 1064–1082. DOI 10.1021/ja00523a026.
- ↑ a b c L. E. Overman; E. J. Jacobsen. The cyanomethyl group for nitrogen protection and iminium ion generation in ring-enlarging pyrrolidine annulations. A short synthesis of the amaryllidaceae alkaloid d,1-crinine. Tetrahedron Letters. 1982, s. 2741–2744. DOI 10.1016/S0040-4039(00)87446-8.
- ↑ a b c d Archivovaná kopie. www.chem.uky.edu [online]. [cit. 2020-12-30]. Dostupné v archivu pořízeném z originálu dne 2015-09-23.
- ↑ a b c S. Jolidon; H. J. Hansen. Untersuchungen über aromatische Amino-Claisen-Umlagerungen. Helvetica Chimica Acta. 1997, s. 978–1032. DOI 10.1002/hlca.19770600329.
- ↑ Ehsan Zahedi; Safa Ali-Asgari; Keley Vahid. NBO and NICS analysis of the allylic rearrangements (the Cope and 3-aza-Cope rearrangements) of hexa-1,5-diene and N-vinylprop-2-en-1-amine: A DFT study. Central European Journal of Chemistry. 2010, s. 1097–1104. DOI 10.2478/s11532-010-0084-1.
- ↑ Kent Brannock; Robert Burpitt. Notes- The Chemistry of Isobutenylamines. II. Alkylation with Allylic and Benzyl Halides. The Journal of Organic Chemistry. 1961, s. 3576–3577. DOI 10.1021/jo01067a64.
- ↑ E. W. Baxter; D. Labaree; H. L. Ammon; P. S. Mariano. Formal total synthesis of deserpidine demonstrating a versatile amino-Claisen rearrangement/Wenkert cyclization strategy for the preparation of functionalized yohimbane ring systems. Journal of the American Chemical Society. 1990, s. 7682–7692. DOI 10.1021/ja00177a032.
- ↑ R. K. Hill; N. W. Gilman. A nitrogen analog of the Claisen rearrangement. Tetrahedron Letters. 1967, s. 1421–1423. DOI 10.1016/S0040-4039(00)71596-6.
- ↑ R. K. Boeckman; M. D. Shair; R. J. Vargas; L. A. Stolz. Synthetic and Mechanistic Studies of the retro-Claisen Rearrangement. 2. A Facile route to Medium-Ring Heterocycles via Rearrangement of Vinylcyclopropane- and Cyclobutanecarboxaldehydes. The Journal of Organic Chemistry. 1993, s. 1295–1297. DOI 10.1021/jo00058a001.
Externí odkazy[editovat | editovat zdroj]
 Obrázky, zvuky či videa k tématu Aza-Copeův přesmyk na Wikimedia Commons
Obrázky, zvuky či videa k tématu Aza-Copeův přesmyk na Wikimedia Commons