
Sigmatropní reakce
Sigmatropní reakce je pericyklická organická reakce, při níž dochází k přeměně jedné vazby σ na jinou vazbu σ při nekatalyzované vnitromolekulární reakci.[1] Jedná se o molekulový přesmyk vedoucí k přesunu substituentu z jedné části π systému na druhou za současného přeuspořádání π systému.[2] Sigmatropní reakce obvykle probíhají za nepřítomnosti katalyzátoru, i když je možná katalýza Lewisovými kyselinami. Nejčastěji prováděnými sigmatropními reakcemi jsou [3,3] Copeův přesmyk, Claisenův přesmyk, Carrollův přesmyk a Fischerova syntéza indolů.
Přehled[editovat | editovat zdroj]
Woodwardovo–Hoffmanovo názvosloví řádů sigmatropních reakcí[editovat | editovat zdroj]
Sigmatropní reakce se popisují pomocí dvojice parametrů [i,j], které jsou definovány tak, že přesunem vazby σ sousedící s jedním nebo více π systémy se na nové místo přesune (i−1) a (j−1) atomů z původní vazby σ.[3] Pokud je i+j sudé číslo, tak jsou do reakce zapojeny neutrální atomy uhlíku, zatímco při liché hodnotě i+j se reakce účastní nabitý atom uhlíku nebo heteroatom s jedním nebo více volnými páry elektronů, který nahrazuje dvojnou vazbu mezi dvěma uhlíky. [1,5] a [3,3] přesmyky se tak za přítomnosti heteroatomu stanou [1,4] a [2,3] přesmyky, aby zůstala zachována symetrie.

Řády sigmatropních reakcí se běžně určují podle počtu atomů tvořících zanikající vazbu a podle počtu atomů vytvářejících novou vazbu σ.[4]

Při přesunech vodíkových atomů se používá podobný postup. Při určení řádu sigmatropního přesmyku zahrnujícího přesuny vodíkových atomů je potřeba počítat přes všechny atomy zapojené do reakce místo pouhého počítání přes sousední atomy, například následující reakce je řádu [1,5] a tento výsledek byl získán počítáním proti směru hodinových ručiček přes π systém, zatímco při počítání přes methylenovou skupinu by vycházel řád [1,3].

Meziprodukt reakce lze určit snadno. U sigmatropní reakce se meziprodukt skládá ze dvou částí, spojených zanikajícími a vznikajícími vazbami σ. Reakce se označuje jako [i,j] přesmyk, pokud tyto části obsahují i a j atomů.

Suprafaciální a antrafaciální přesmyky[editovat | editovat zdroj]
Při sigmatropních reakcích může být stereochemie přesunující se skupiny zachována, ale může se také obrátit, podle toho, zda novou vazbu vytvoří stejný lalok orbitalu přesunujícího se atomu, jaký vytvářel původní vazbu, nebo bude tato vazba tvořena překryvem jiného laloku.[4]

Pokud zůstane stereochemie zachována, tak se skupina přesune, aniž by došlo k rotaci kolem vazby, která způsobuje obrácení stereochemie.
Na stereochemii produktu může ovšem vliv také to, jestli po vzniku nové vazby zůstane přesunující se skupina na stejné straně π systému. Pokud tomu tak je, jedná se o suprafaciální přesmyk, v opačném případě jde o antarafaciální přesmyk.[3] Antarafaciální přesmyky nemohou probíhat u příliš malých cyklů.

Druhy sigmatropních přesmyků[editovat | editovat zdroj]
[1,3] přesmyky[editovat | editovat zdroj]
Tepelné přesmyky hydridů[editovat | editovat zdroj]
Při tepelných [1,3] přesmycích hydridů dojde k přesunu tří atomů na molekule hydridu. Podle Woodwardových–Hoffmannových pravidel probíhají tyto reakce antarafaciálně. I když takový posun dovoluje symetrie, tak jej znemožňuje Möbiova topologie příslušného meziproduktu; díky němu u enolů nedochází k izomerizaci, pokud není přítomna kyselina nebo zásada.[4]

K tomuto přesmyku nemůže dojít.

Tepelné přesmyky alkylů[editovat | editovat zdroj]
[1,3] přesmyky alkylových skupin, podobné [1,3] přesmykům hydridů, mohou probíhat pouze antarafaciálně. Geometrie meziproduktu reakci znesnadňuje, ovšem alkylové skupiny, díky vlastnostem svých orbitalů, mohou převrátit svou geometrii, čímž vytvoří novou vazbu pomocí zadního laloku sp3 orbitalu, což vede k suprafaciálnímu přesmyku. Tyto reakce neprobíhají běžně u sloučenin s acyklickými řetězci, protože příslušný meziprodukt snadněji vzniká z cyklických sloučenin.[4]
![[1,3] přesuny alkylových skupin](//upload.wikimedia.org/wikipedia/commons/thumb/e/e9/1%2C3alkylfixed.png/550px-1%2C3alkylfixed.png)
[1,3] přesuny alkylových skupin
![[1,3] přesuny alkylových skupin](/wiki/Soubor:1,3alkylfixed.png)
Fotochemické [1,3] přesmyky[editovat | editovat zdroj]
Fotochemické [1,3] přesmyky by měly probíhat suprafaciálně, většinou však nemají soustředěný mechanismus, protože probíhají přes tripletové meziprodukty, tedy diradikálovým mechanismem, na který se Woodwardova–Hoffmannova pravidla nevztahují.
[1,5] přesmyky[editovat | editovat zdroj]
[1,5] přesmyky spočívají v přesunu jednoho substituentu (vodíku, alkylové, nebo arylové skupiny) přes 5 atomů π systému. K přesunu vodíků dochází obvykle při teplotách nad 200 °C.[4] Tyto reakce probíhají většinou suprafaciálně přes meziprodukt s Hückelovou topologií.
![[1,5] přesun vodíku v cyklickém π systému](//upload.wikimedia.org/wikipedia/commons/thumb/7/7f/1%2C5hydridecyclicfixed.png/300px-1%2C5hydridecyclicfixed.png)
[1,5] přesun vodíku v cyklickém π systému
![[1,5] přesun vodíku v cyklickém π systému](/wiki/Soubor:1,5hydridecyclicfixed.png)
I když se antarafaciální [1,5] přesmyky objevují vzácně, tak je také lze provést a byly popsány případy, kdy antarafaciální mechanismus převažoval[5]
![Antarafaciální [1,5] přesmyk](//upload.wikimedia.org/wikipedia/commons/thumb/9/99/1%2C5hantarafacialfixed.png/600px-1%2C5hantarafacialfixed.png)
Antarafaciální [1,5] přesmyk
![Antarafaciální [1,5] přesmyk](/wiki/Soubor:1,5hantarafacialfixed.png)
Na rozdíl od vodíkových přesmyků nebyly nikdy pozorovány [1,5] alkylové přesmyky u acyklických sloučenin;[4] existují ovšem studie, ze kterých vyplývá, že podle druhu substituentu klesá rychlost [1,5] přesmyků u cyklických sloučenin v řadě karbonyl a karboxyl > hydrid > fenyl a vinyl (ethenyl) >> alkyl.[6][7]
U alkylových skupin probíhají [1,5] přesmyky obtížně, často pouze za velmi vysokých teplot, ale u derivátů cyklohexa-1,3-dienu k nim dochází i při nižších teplotách než u karbonylových derivátů, které jsou nejlepšími přesunujícími se skupinami. Nízká reaktivita alkylových skupin je způsobena tím, že reakce probíhá jiným mechanismem, kdy nejprve dojde k otevření cyklu, po němž následuje [1,7] přesmyk a následně proběhne elektrocyklické uzavření kruhu.:[8]

Přesun alkylové skupiny u derivátu cyklohexadienu

Níže je zobrazena podobná reakce, pouze bez obnovení cyklu, kterou se přeměňuje lumisterol na vitamin D2.
[1,7] přesmyky[editovat | editovat zdroj]
[1,7] sigmatropní přesmyky by podle Woodwardových–Hoffmannových pravidel měly probíhat antarafaciálně. Při přeměně lumisterolu na vitamin D2 byl pozorován antarafaciální [1,7] přesmyk methylové skupiny následující po otevření cyklu provitaminu D2.[9]

Přeměna lumisterolu na vitamin D2

U bicyklických nonatrienů dochází k [1,7] přesmykům,,[10] při kterých se přesune dvojvazná skupina v tříčlenném cyklu bicyklické molekuly.
![]1,7] přesmyk u bicyklického nonatrienu](//upload.wikimedia.org/wikipedia/commons/thumb/c/ce/1%2C7walknonatriene.png/300px-1%2C7walknonatriene.png)
]1,7] přesmyk u bicyklického nonatrienu
![]1,7] přesmyk u bicyklického nonatrienu](/wiki/Soubor:1,7walknonatriene.png)
[3,3] přesmyky[editovat | editovat zdroj]
Nejlépe prozkoumané jsou [3,3] sigmatropní přesmyky. Podle Woodwardových–Hoffmannových pravidel probíhají suprafaciálně.
Claisenovy přesmyky[editovat | editovat zdroj]
Claisenovy přesmyky objevil v roce 1912 Rainer Ludwig Claisen; jedná se o první popsaný druh [3,3] sigmatropního přesmyku.[11][12][13] Jedná se o významný způsob tvorby vazeb uhlík-uhlík. Příkladem může být [3,3] přesmyk allylvinyletheru, ze kterého při zahřátí vzniká γ,δ-nenasycená karbonylová sloučenina. Tvorba karbonylové skupiny způsobuje, že je tato reakce, na rozdíl od většiny sigmatropních reakcí, nevratná.

Claisenův přesmyk

Aromatické Claisenovy přesmyky[editovat | editovat zdroj]
Při ortho-Claisenových přesmycích probíhá [3,3] přesun na molekule allylfenyletheru, přičemž se tvoří meziprodukt, který se následně tautomerizuje na ortho-substituovaný fenol.
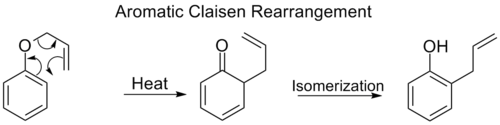
Aromatický Claisenův přesmyk

Pokud jsou obě ortho polohy blokovány, tak proběhne para-Claisenův přesmyk a produktem následné tautomerizace bude trisubstituovaný fenol:

Para-Claisenův přesmyk

Copeův přesmyk[editovat | editovat zdroj]
Copeův přesmyk je často zkoumanou reakcí, jedná se o [3,3] sigmatropní přesmyk u 1,5-dienů.[14][15][16] Objevil jej Arthur Clay Cope. Přikladem této reakce je přeměna 3,4-dimethyl-hexa-1,5-dienu za teploty 300 °C na okta-2,6-dien.

Copeův přesmyk 3,4-dimethyl-hexa-1,5-dienu

Oxy-Copeův přesmyk[editovat | editovat zdroj]
Při oxy-Copeově přesmyku se připojí hydroxylová skupina na C3 za vzniku nenasycené karbonylové sloučeniny (enalu nebo enonu) vytvořeného tautomerizací enolového meziproduktu:[17]
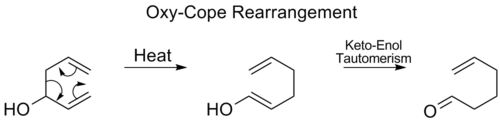
Oxy-Copeův přesmyk

Carrollův přesmyk[editovat | editovat zdroj]
Carrollův přesmyk je přeměna β-keto-allylového esteru na α-allyl-β-ketokarboxylovou kyselinu[18] spojená s následnou dekarboxylací za vzniku γ,δ-allylketonu. Jedná se o variantu Claisenova přesmyku a v podstatě o dekarboxylační allylaci.

Carrollův přesmyk

Fischerova syntéza indolů[editovat | editovat zdroj]
Fischerova syntéza indolů slouží k přípravě indolů ze substituovaných fenylhydrazinů a aldehydů nebo ketonů v kyselém prostředí.[19][20] Objevil ji Hermann Emil Fischer v roce 1883.

Fischerova syntéza indolů

Velký význam má výběr konkrétní kyseliny použité ke katalýze. Brønstedovy kyseliny jako HCl, H2SO4, kyselina fosforečná a kyselina p-toluensulfonová jsou vhodné pro tuto reakci, podobně lze použít i Lewisovy kyseliny, jako fluorid boritý, chlorid zinečnatý , chlorid železitý a chlorid hlinitý.
Tento druh reakce byl několikrát zkoumán.[21][22][23]
[5,5] přesmyky[editovat | editovat zdroj]
Podobně jako [3,3] přesmyky probíhají i [5,5] přesmyky suprafaciálně. Jsou méně časté než [3,3] přesmyky, což je ovšem způsobeno hlavně menším počtem prozkoumaných molekul, u kterých takové reakce mohou probíhat.[4]
![[5,5] přesmyk fenylpentadienyletheru](//upload.wikimedia.org/wikipedia/commons/thumb/2/25/5%2C5shiftfixeds.png/800px-5%2C5shiftfixeds.png)
[5,5] přesmyk fenylpentadienyletheru
![[5,5] přesmyk fenylpentadienyletheru](/wiki/Soubor:5,5shiftfixeds.png)
[2,3] přesmyky[editovat | editovat zdroj]
Mezi [2,3]-sigmatropní přesmyky patří například [2,3]-Wittigův přesmyk.
Odkazy[editovat | editovat zdroj]
Související články[editovat | editovat zdroj]
Reference[editovat | editovat zdroj]
V tomto článku byl použit překlad textu z článku Sigmatropic reaction na anglické Wikipedii.
- ↑ Carey, F.A. and R.J. Sundberg. Advanced Organic Chemistry Part A ISBN 0-306-41198-9
- ↑ Sigmatropic Rearrangements [online]. Dostupné online.
- ↑ a b Robert Burns Woodward; Roald Hoffmann The Conservation of Orbital Symmetry. Verlag Chemie Academic Press. 2004. ISBN 0-89573-109-6.
- ↑ a b c d e f g Miller, Bernard. Advanced Organic Chemistry. 2nd Ed. Upper Saddle River: Pearson Prentice Hall. 2004. ISBN 0-13-065588-0
- ↑ Kiefer, E.F.; Tana, C.H. Journal of the American Chemical Society, 1969, 91, 4478. DOI:10.1021/ja01044a027
- ↑ Fields, D.J.; Jones, D.W.; Kneen, G. Chemical Communications 1976. 873 – 874. DOI:10.1039/C39760000873
- ↑ Miller, L.L.; Greisinger, R.; Boyer, R.F. J. Am. Chem. Soc. 1969. 91. 1578. DOI:10.1021/ja01034a076
- ↑ Schiess, P.; Dinkel, R. Tetrahedron Letters, 1975, 16, 29, 2503. DOI:10.1016/0040-4039(75)80050-5
- ↑ Francis A. Carey; Richard J. Sundberg. Advanced Organic Chemistry. Part A: Structure and Mechanisms. New York: Kluwer Academic/Plenum, 2000. Dostupné online. ISBN 0-306-46242-7. S. 625.
- ↑ Klaerner, F.G. Angewandte Chemie International Edition in English, 1972, 11, 832.DOI:10.1002/anie.197208321
- ↑ Claisen, L.; Ber. 1912, 45, 3157. DOI:10.1002/cber.19120450348
- ↑ Claisen, L.; Tietze, E.; Chemische Berichte 1925, 58, 275. DOI:10.1002/cber.19250580207
- ↑ Claisen, L.; Tietze, E.; Chemische Berichte 1926, 59, 2344. DOI:10.1002/cber.19260590927
- ↑ Cope, A. C.; et al. J. Am. Chem. Soc. 1940, 62, 441. DOI:10.1021/ja01859a055
- ↑ Hoffmann, R.; Stohrer, W. D. J. Am. Chem. Soc. 1971, 93, 25, 6941–6948. DOI:10.1021/ja00754a042
- ↑ Dupuis, M.; Murray, C.; Davidson, E. R. J. Am. Chem. Soc. 1991, 113, 26, 9756–9759. DOI:10.1021/ja00026a007
- ↑ Berson, Jerome A.; Jones, Maitland. J. Am. Chem. Soc. 1964, 86, 22, 5019–5020. DOI:10.1021/ja01076a067
- ↑ Carrol, M. F. Journal of the Chemical Society 1940, 704–706. DOI:10.1039/JR9400000704.
- ↑ Fischer, E.; Jourdan, F. Chemische Berichte 1883, 16, 2241.DOI:10.1002/cber.188301602141
- ↑ Fischer, E.; Hess, O. Chemische Berichte 1884, 17, 559. DOI:10.1002/cber.188401701155
- ↑ van Orden, R. B.; Lindwell, H. G. Chem. Rev. 1942, 30, 69–96. DOI:10.1021/cr60095a004
- ↑ Robinson, B. Chem. Rev. 1963, 63, 373–401. DOI:10.1021/cr60224a003
- ↑ Robinson, B. Chem. Rev. 1969, 69, 227–250. DOI:10.1021/cr60262a003